
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2017
Volume: 7, Issue: 11

Biochemistry

Heavy Metal Stress Assay of Caenorhabditis elegans
Cancer Biology

DNA Fiber Assay upon Treatment with Ultraviolet Radiations



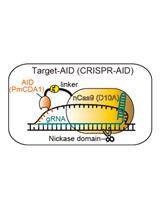
Targeted Nucleotide Substitution in Mammalian Cell by Target-AID
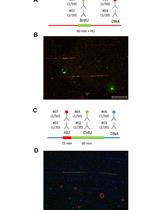
Single-molecule Analysis of DNA Replication Dynamics in Budding Yeast and Human Cells by DNA Combing

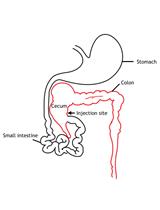
Intracaecal Orthotopic Colorectal Cancer Xenograft Mouse Model

Fluorometric Estimation of Glutathione in Cultured Microglial Cell Lysate





Pituitary Isograft Transplantation in Mice




Whole Mammary Gland Transplantation in Mice Protocol

Cell Biology

Generation of Mutant Pigs by Direct Pronuclear Microinjection of CRISPR/Cas9 Plasmid Vectors



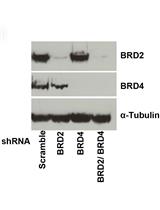
Flow Cytometric Analysis of HIV-1 Transcriptional Activity in Response to shRNA Knockdown in A2 and A72 J-Lat Cell Lines
Immunology
ELISPOT Assay to Measure Peptide-specific IFN-γ Production


In vitro Antigen-presentation Assay for Self- and Microbial-derived Antigens

Isolation and Infection of Drosophila Primary Hemocytes
Microbiology
Expression and Purification of the Cas10-Csm Complex from Staphylococci



A Reliable Assay to Evaluate the Virulence of Aspergillus nidulans Using the Alternative Animal Model Galleria mellonella (Lepidoptera)

Fluorescently Labelled Aerolysin (FLAER) Labelling of Candida albicans Cells

Phototaxis Assays of Synechocystis sp. PCC 6803 at Macroscopic and Microscopic Scales
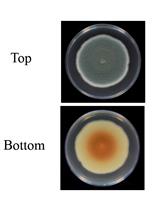
Induction and Quantification of Patulin Production in Penicillium Species
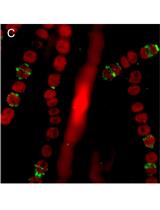
Protein Localization in the Cyanobacterium Anabaena sp. PCC7120 Using Immunofluorescence Labeling



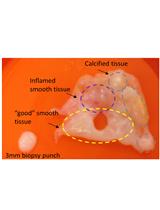
Ex vivo Model of Human Aortic Valve Bacterial Colonization



Molecular Biology
Chromatin Immunoprecipitation Experiments from Whole Drosophila Embryos or Larval Imaginal Discs

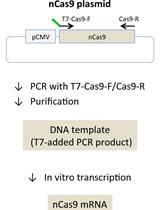
A Protocol for Production of Mutant Mice Using Chemically Synthesized crRNA/tracrRNA with Cas9 Nickase and FokI-dCas9
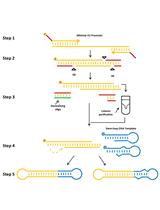
Formation of Minimised Hairpin Template-transcribing Dumbbell Vectors for Small RNA Expression


Neuroscience

Kinetic Lactate Dehydrogenase Assay for Detection of Cell Damage in Primary Neuronal Cell Cultures

The Repeated Flurothyl Seizure Model in Mice

Plant Science
DNA-free Genome Editing of Chlamydomonas reinhardtii Using CRISPR and Subsequent Mutant Analysis

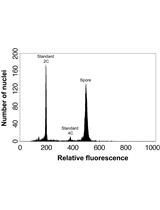
Determining Genome Size from Spores of Seedless Vascular Plants

Protocol for Enrichment of the Membrane Proteome of Mature Tomato Pollen




Photometric Assays for Chloroplast Movement Responses to Blue Light
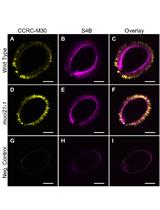
Whole-seed Immunolabeling of Arabidopsis Mucilage Polysaccharides
Stem Cell
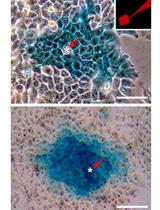
Functional Analysis of Connexin Channels in Cultured Cells by Neurobiotin Injection and Visualization

