
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2020
Volume: 10, Issue: 19
Biophysics

Assembly and Imaging Set up of PIE-Scope
Cancer Biology

A Novel and Robust Single-cell Trapping Method on Digital Microfluidics


Cell Biology
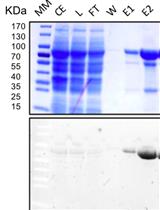
Affinity Purification of GO-Matryoshka Biosensors from E. coli for Quantitative Ratiometric Fluorescence Analyses

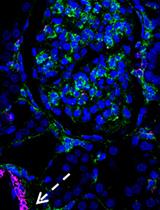
Double Labeling of PDGFR-β and α-SMA in Swine Models of Acute Kidney Injury to Detect Pericyte-to-Myofibroblast Transdifferentation as Early Marker of Fibrosis

Developmental Biology

Fluorescence Measurement and Calibration of Intracellular pH in Starfish Oocytes

Microbiology
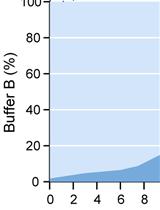
Analysis of Gram-negative Bacteria Peptidoglycan by Ultra-performance Liquid Chromatography
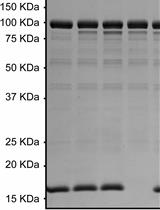
Radioactive Assay of in vitro Glutamylation Activity of the Legionella pneumophila Effector Protein SidJ

Simple Time-lapse Imaging for Quantifying the Hydrostatic Production of Oxygenic Photogranules




TetR Regulated in vivo Repression Technology to Identify Conditional Gene Silencing in Genetically Engineerable Bacteria Using Vibrio cholerae Murine Infections as Model System
Molecular Biology
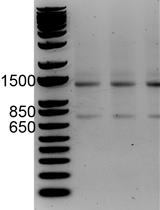
Using RNA Sequencing and Spike-in RNAs to Measure Intracellular Abundance of lncRNAs and mRNAs

Neuroscience

An Operant Conditioning Model Combined with a Chemogenetic Approach to Study the Neurobiology of Food Addiction in Mice
Preparing Viable Hippocampal Slices from Adult Mice for the Study of Sharp Wave-ripples

Plant Science
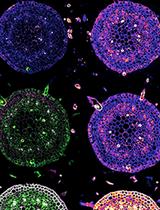
Multitarget Immunohistochemistry for Confocal and Super-resolution Imaging of Plant Cell Wall Polysaccharides

A Protocol for Flavonols, Kaempferol and Quercetin, Staining in Plant Root Tips
Stem Cell
Fluidigm Based Single-cell Gene Expression Library Preparation from Patient-derived Small Intestinal Organoids