
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2020
Volume: 10, Issue: 20

Cancer Biology

Quantification of Salivary Charged Metabolites Using Capillary Electrophoresis Time-of-flight-mass Spectrometry


Real-time Three-dimensional Tracking of Endocytic Vesicles
Cell Biology

Live-cell Imaging and Quantitative Analysis of Meiotic Divisions in Caenorhabditis elegans Males
Microbiology
A Protocol for Simple, Rapid, and Direct Detection of SARS-CoV-2 from clinical samples, using Reverse Transcribed Loop-Mediated Isothermal Amplification (RT-LAMP)
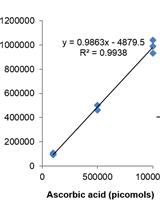
Measurement of Ascorbic Acid and Glutathione Content in Cyanobacterium Synechocystis sp. PCC 6803


Estimation of the Minimum Number of Replication Origins Per Chromosome in any Organism

Molecular Biology
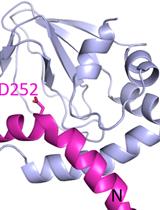
Understanding Docking Complexes of Macromolecules Using HADDOCK: The Synergy between Experimental Data and Computations
Attachment of a 32P-phosphate to the 3′ Terminus of a DNA Oligonucleotide
Analyzing (Re)Capping of mRNA Using Transcript Specific 5' End Sequencing
Neuroscience
Staining and Quantitative Analysis of Myelinating Oligodendrocytes in the Mouse Grey Matter
Plant Science
Low-cost and High-throughput RNA-seq Library Preparation for Illumina Sequencing from Plant Tissue



Dual sgRNA-based Targeted Deletion of Large Genomic Regions and Isolation of Heritable Cas9-free Mutants in Arabidopsis

Acidified Blue Ink-staining Procedure for the Observation of Fungal Structures Inside Roots of Two Disparate Plant Lineages


.JPEG)

Quantification of Methylglyoxal Levels in Cowpea Leaves in Response to Cowpea Aphid Infestation
Stem Cell
Derivation of Induced Pluripotent Stem Cells from Human Fibroblasts Using a Non-integrative System in Feeder-free Conditions