
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2022
Volume: 12, Issue: 9

Biochemistry
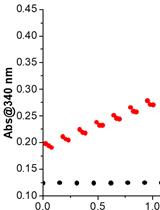
Expression, Purification, and in vitro Enzyme Activity Assay of a Recombinant Aldehyde Dehydrogenase from Thermus thermophilus, using an Escherichia coli host
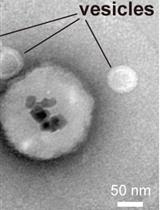
Immunoisolation of Endosomal Recycling Vesicles from Saccharomyces cerevisiae
Biophysics
Production and Crystallization of Nanobodies in Complex with the Receptor Binding Domain of the SARS-CoV-2 Spike Protein
Cell Biology
Quantitative Analysis of Actin Cable Length in Yeast
Computational Biology and Bioinformatics

A System to Easily Manage Metadata in Biomedical Research Labs Based on Open-source Software
Microbiology
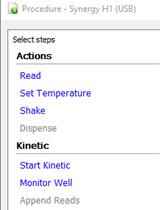
Bacterial Growth Curve Measurements with a Multimode Microplate Reader
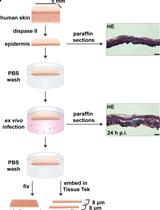
Ex vivo Human Skin Infection with Herpes Simplex Virus 1
Neuroscience
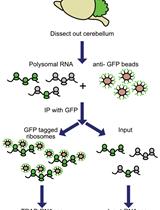
Translating Ribosome Affinity Purification (TRAP) of Cell Type-specific mRNA from Mouse Brain Lysates
Plant Science

Quantification of Soil-surface Roots in Seedlings and Mature Rice Plants
Systems Biology
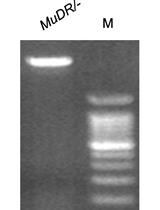
A Molecular Cloning and Sanger Sequencing-based Protocol for Detecting Site-specific DNA Methylation