
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2023
Volume: 13, Issue: 21
Biological Engineering
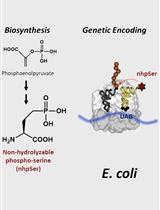
Biosynthesis and Genetic Encoding of Non-hydrolyzable Phosphoserine into Recombinant Proteins in Escherichia coli
Cancer Biology
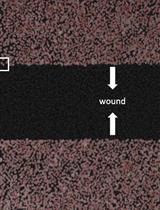
Studying Cell Migration (Random and Wound Healing) Parameters with Imaging and MATLAB Analysis
Cell Biology
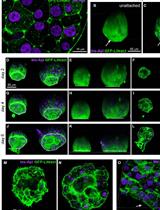
Preparation of Whole-mount Mouse Islets on Vascular Extracellular Matrix for Live Islet Cell Microscopy
Identification of Acetylation Sites of Fatty Acid Synthase (FASN) by Mass Spectrometry and FASN Activity Assay
Drug Discovery
Studying Cellular Focal Adhesion Parameters with Imaging and MATLAB Analysis
Immunology
Medullary Thymic Epithelial Cell Antigen-presentation Assays
Microbiology
Analysis of Plasmodium falciparum Mitochondrial Electron Transport Chain Activity Using Seahorse XFe96 Extracellular Flux Assays
Detailed Protocol to Perform Direct PCR Using Filamentous Fungal Biomass—Tips and Considerations
A Guideline for Assessment and Characterization of Bacterial Biofilm Formation in the Presence of Inhibitory Compounds
Spot Assay and Colony Forming Unit (CFU) Analyses–based sensitivity test for Candida albicans and Saccharomyces cerevisiae
Molecular Biology
Purification of Long Non-coding RNAs on Replication Forks Using iROND (Isolate RNAs on Nascent DNA)
Neuroscience
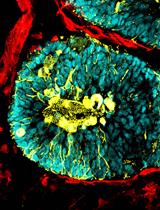
Generation of Human Blood Vessel and Vascularized Cerebral Organoids
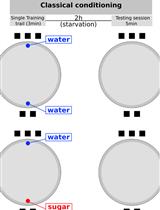
A New Behavioral Paradigm for Visual Classical Conditioning in Drosophila
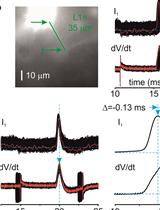
Measuring Action Potential Propagation Velocity in Murine Cortical Axons
Stem Cell

Differentiation of Human Induced Pluripotent Stem Cells (iPSCs)–derived Mesenchymal Progenitors into Chondrocytes
Systems Biology

Workflow for High-throughput Screening of Enzyme Mutant Libraries Using Matrix-assisted Laser Desorption/Ionization Mass Spectrometry Analysis of Escherichia coli Colonies