
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

U2.3 Precursor Small Nuclear RNA in vitro Processing Assay
(*contributed equally to this work) Published: Vol 11, Iss 17, Sep 5, 2021 DOI: 10.21769/BioProtoc.4142 Views: 1363
Reviewed by: Zhibing LaiSwati MeghaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics



Abstract
Small nuclear RNAs (snRNAs) are vital for eukaryotic cell activities and play important roles in pre-mRNA splicing. The molecular mechanism underlying the transcription of snRNA, regulated via upstream/downstream cis-elements and relevant trans-elements, has been investigated in detail using cell-free extracts. However, the processing of precursor snRNA (pre-snRNA), which is required by 3’ end maturation of pre-snRNA, remains unclear as a proper processing assay is difficult to develop in vitro. Here, we present an in vitro method using synthetic labeled RNA as substrates to study the 3’ cleavage of pre-snRNA.
Keywords: snRNABackground
As critical components of spliceosome for the processing and splicing of precursor mRNAs (pre-mRNAs), small nuclear RNAs (snRNAs) are essential and fundamental non-coding small RNAs in eukaryotic cells. The transcription complex relying on the DNA-dependent RNA polymerase II (Pol II) is required for most snRNA transcription (including U1, U2, U4, and U5), except for U6, which depends on RNA polymerase III (Pol III) (Carbon et al., 1987; Vankan and Filipowicz, 1988). The Pol II mediated transcription of snRNA requires a set of integrator factors (INTs) in metazoans (Baillat et al., 2005). INTs contain at least 14 subunits in metazoans (Baillat et al., 2005; Chen and Wagner, 2010), associated with C-terminal domain (CTD) of Pol II to form the transcription complex. Recently, five plant INT factors involved in snRNA biosynthesis were identified in Arabidopsis and termed Defective in snRNA Processing (DSP), including DSP1-4 and CPSF73-I (Cleavage and Polyadenylation Specificity Factor 73 kDa) (Liu et al., 2016). The INTs/DSPs are recruited to the promoter of snRNA and associate with Pol II to synthesize the pre-snRNA, transcribing it beyond the 3’ end of mature snRNAs. The nascent pre-snRNA transcripts contain the snRNA sequence and the excessive 3’ box RNA fragment. Consequently, pre-snRNAs are subject to 3’ maturation, a process involving endonucleolytic cleavage of the nascent transcript at the cleavage site located on the upstream of the 3’ box, to produce mature snRNA (Uguen and Murphy, 2003 and 2004; Baillat et al., 2005; Chen and Wagner, 2010).
The molecular mechanism underlying the initiation and inhibition of snRNA transcription has been well studied both in vivo and in vitro. The functions of cis-elements, including the distal sequence element (DSE), proximal sequence element (PSE) in humans and upstream sequence element (USE) in plants, and trans-elements, including INTs/DSPs, have been investigated in detail (Hernandez, 2001).
Compared with the well-studied regulation of snRNA transcription, the processing steps of pre-snRNA remain poorly understood (Hernandez, 2001; Jawdekar and Henry, 2008). Most studies on pre-snRNA processing were performed by evaluating the relative content of pre-snRNA in specific genotype material. An efficient system for detecting the processing of pre-snRNA using cell extracts will facilitate the study of the maturation of pre-snRNA in specific genotypic plants and reveal the influence of specific factors on pre-RNA maturation after transcription.
Accordingly, we applied a synthetic pre-U2.3 snRNA and created a system to analyze the in vitro processing activities of pre-U2.3 using the production of cell extracts. This in vitro processing analysis provides a method to detect the activation of pre-snRNA maturity.
Materials and Reagents
Nylon membrane (Invitrogen, catalog number: LC2003)
DNaseI (NEB, catalog number: M0303S)
Nuclease-free water (ThermoFisher Scientific, catalog number: AM9932)
Trizol (ThermoFisher Scientific, catalog number: 10296028)
Chloroform (Sigma-Aldrich, catalog number: 288306)
Isopropanol (Sigma-Aldrich, catalog number: 563935)
Ethanol (Sigma-Aldrich, catalog number: 51976)
2-mercaptoethanol (Gibco, catalog number: 21985023)
Hexylene glycol (Sigma-Aldrich, catalog number: 112100)
Triton X-100 (Fisher Scientific, Acros organics, catalog number: 215680010)
dNTP (10 mM each) (ThermoFisher Scientific, catalog number: R0192)
NTP (10 mM each) (ThermoFisher Scientific, Molecular biology grade NTPs, catalog number: R0481)
Glycerol (Sigma-Aldrich, catalog number: G5516)
HEPES (Sigma-Aldrich, catalog number: H3375)
Various salts: NaCl, KCl, MgCl2, and MnCl2 (Sigma-Aldrich, catalog numbers: S9888, P9541, M8266, and M1787, respectively)
Creatine phosphate (Sigma-Aldrich, catalog number: 10621714001)
Polyvinyl alcohol (Sigma-Aldrich, catalog number: 341584)
DTT (ThermoFisher Scientific, catalog number: R0861)
Pepstatin A (Sigma-Aldrich, catalog number: P5318)
Phenylmethylsulfonyl (PMSF) (Sigma-Aldrich, catalog number: 52332)
Protease inhibitor (Roche, catalog number:11697498001)
Diethyl pyrocarbonate (DEPC) (Sigma-Aldrich, catalog number: D5758)
[γ-32P]-ATP (3,000 Ci/mmol, 10 mCi/ml) (PerkinElmer, catalog number: BLU002A)
T7 RNA polymerase (ThermoFisher, catalog number: 18033019)
T4 polynucleotide kinase (PNK) (NEB, catalog number: M0201S)
High-fidelity DNA polymerase (ThermoFisher, catalog number: F530L)
pCR-Blunt (ThermoFisher, catalog number: K270020)
A map of pCR-Blunt can be found on the website of the manufacturer: www.thermofisher.com/order/catalog/product/K270020#/K270020.
RNA marker (Abnova, catalog number: R0002)
Bradford reagent (Bio-Rad, catalog number: 5000205)
Gel Purification kit
PCR Extraction Kit
RNase away
Primers:
U2.3-RNA-F: atacctttctcggccttttggc
U2.3-RNA-R: ctgcgtaacatatataaatatctctg
T7-U2.3-F: TAATACGACTCACTATAGGGatacctttctcggc
PolyG-U2.3-R: CCCCCCCCCCCCctgcgtaacatatataa
Processing buffer (see Recipes)
M1 buffer (see Recipes)
M2 buffer (see Recipes)
M3 buffer (see Recipes)
2× formamide loading buffer (see Recipes)
6% denaturing polyacrylamide gel containing 6 M urea (see Recipes)
Equipment
PhosphorImager (GE Health Care, model: Typhoon FLA 9500)
Suitable space for working with 32P radioactivity
Geiger counter (Thermo, 900 mini)
Plexiglass shield to protect user from radioactivity
Plexiglass box for 32P waste
Sephadex G-25 spin column (Merck, 11273990001)
PCR Machine
Phosphor Screen (Molecular Dynamics)
Cell strainer (Biofil, catalog number: CSS013100)
Vertical electrophoresis gel box and glass plates with spacers and combs (Bio-Rad, model: Mini-PROTEAN Tetra)
Spectrophotometer (any UV absorption spectrophotometry including Bradford available)
Software
Quantity One (Bio-Rad Laboratories) or ImageJ
Procedure
Assemble pre-U2.3 templates
Add the following to the PCR reaction:
36.5 μl ddH2O
10 μl 5× Phusion HF buffer
1 μl 10 mM dNTP mix
200 ng DNA of Arabidopsis
0.5 μl U2.3-RNA-F and U2.3-RNA-R primers (10 pmol/μl)
0.5 μl High-Fidelity DNA Polymerase
Perform PCR to generate pre-U2.3 fragment with the following cycles:
98°C for 30 s to denature the DNA
95°C for 10 s
58°C for 30 s
72°C for 15 s
Repetition of PCR for 31 cycles
72°C for 10 min
4°C for storage
Electrophorese PCR-generated DNA fragments in 1.5% agarose gel.
Verify the pre-U2.3 (355 bp) fragment and purify PCR product from agarose gel using Gel Purification kit following manufacturer’s instructions.
Cloning of the amplified PCR products to pCR-Blunt.
6 μl purified pre-U2.3 PCR product
1 μl pCR-Blunt vector
2 μl 5× T4 DNA Ligase buffer
1 μl T4 DNA Ligase
Incubate the ligation reaction at room temperature for 10 min. Transform the construct into competent Escherichia coli. Extract the positive vector and confirm the pre-U2.3 fragment using singer sequencing.
Assemble pre-U2.3 templates and fuse with T7 promoter and Ploy-G. Add the following to the reaction:
74 μl ddH2O
20 μl 5× Phusion HF buffer
2 μl 10 mM dNTP mix
1 μl pre-U2.3 pCR-Blunt vector (50 ng/μl)
1 μl T7-U2.3-F and PolyG-U2.3-R primers (10 pmol/μl)
1 μl High-Fidelity DNA Polymerase
Perform PCR with the following cycles:
98°C for 30 s to denature the DNA
95°C for 10 s
58°C for 30 s
72°C for 15 s
Repetition of PCR for 33 cycles
72°C for 10 min
4°C for storage
Load a small aliquot of PCR product (5 μl) in 1.5% agarose gel for checking the quality of PCR fragments. If the product contains a single band, the rest of the product (95 μl) can be purified directly using the PCR Extraction Kit without gel purification.
Elute the PCR product in the final 50 μl DEPC water, used as in vitro transcription template.
Synthesize pre-U2.3-polyG snRNA substrates
In vitro RNA transcription. Add the following to the reaction:
22 μl DEPC water
10 μl 5× Transcription Buffer (Ambion)
2.5 μl NTPs (10 mM ATP, CTP, UTP, GTP)
1 μl RNasin (Promega 40 units/μl)
1 μl 1 M DTT
5 μl Transcription Template (200 ng/μl)
1 μl T7 RNA Polymerase (50 unit/μl)
Incubate for 1 h at 37°C
Add 1 μl of DNase I (10 units/μl) and incubate for 15 min at 37°C.
Add 50 μl 2× formamide loading buffer to the reaction solution, boil for 5 min, and load onto a pre-run 5% denaturing polyacrylamide gel containing 6 M urea. Separate RNAs at 20 mA/gel, ~1 h.
After electrophoresis, visualize the PAGE gel with ethidium bromide and cut out the RNA bands directly with a clean razor blade. The gel slice is crushed into a fine slurry, which is soaked in the TE buffer. The slurry is then centrifuged and the supernatant recovered with a pipette. Polyacrylamide fragments carried over into the supernatant can be removed by filtration through a 0.2 mm filter.
Add 3 M sodium acetate (pH 5.2) to a final concentration of 0.3 M and use 3 volumes of absolute ethanol to precipitate RNA. Pellet the RNA by centrifuging at 12,000 × g for 15 min.
Wash small pellets with 75% ethanol to remove undesired salts.
Dissolve the RNA substrates in 20 μl DEPC water.
Radioactive labeling of RNA substrates. Add the following to the reaction:
3 μl DEPC H2O
20 μl RNA substrate
3 µl T4 PNK Reaction Buffer (10×)
1.5 µl T4 PNK
1.5 μl RNasin (Promega 40 units/µl)
1 µl [γ-32P]-ATP
Incubate at 37°C for 30 min. Add 30 μl DEPC water (This volume can be varied if more or less pre-snRNA probe is desired).
Unincorporated nucleotides are removed using a Sephadex G-25 quick spin column.
Synthesize radioactive RNA marker
Take 15 μl commercial RNA marker into Sephadex G-25 column to remove the salt and dye.
For radioactive labeling of the RNA marker, add the following to the reaction:
10 μl filtered RNA marker
6 μl DEPC water
2 µl T4 PNK Reaction Buffer (10×)
1 µl T4 PNK
1 µl [γ-32P]-ATP
Incubate at 37°C for 30 min
Remove unincorporated nucleotides using a Sephadex G-25 quick spin column.
Prepare a dilution series of the labeled RNA marker (1:5, 1:15, 1:50, and 1:150). Drip 2 µl of series diluted marker and pre-U2.3-polyG RNA substrate onto the nylon membrane. Fix the RNAs spot to the membrane by cross linking using the UV crosslinker with a setting of 120,000 µJ cm-2.
Place the plastic-wrapped nylon membrane on the storage phosphor imaging screen and expose for 8 h. The phosphor screen is then scanned with PhosphorImager.
Dilute RNA marker using RNA loading buffer to same radiation level with pre-U2.3-polyG RNA substrate according to the radioactive signal of the series dilution spot.
In vitro processing assay of pre-U2.3-polyG snRNA
Carefully grind 2 g inflorescence to fine powder in liquid N2 and pour the powder into 15 ml tube.
Set the tube on ice, add 8 ml M1 buffer, and invert several times.
Filter the solution through cell strainer (100 µM) into a 50 ml tube.
Centrifuge at 12,000 × g for 10 min at 4°C.
Discard the supernatant, add 4 ml M2 buffer, and mix by pipetting. Transfer the solution into a 5 ml tube. Wash with 3 ml M2 buffer and 3 ml M3 buffer, consecutively, each wash by spin 1 min at 13,200 × g. Remove all of the solution with a small tip pipette.
Add 200 μl processing buffer (without creatine phosphate) to suspend the pellet.
Take 10 μl nuclear protein into 190 μl Bradford reagent, let stand for 5 min, and measure the protein concentrations at 595 nm.
Dilute the nuclear protein of each sample to 50 ng/μl using processing buffer (without creatine phosphate).
For a total of 50 μl reaction volume for each repeat, incubate 40 μl of nuclear protein, 5 μl of labeled DNA, and 5 μl of 200 mM creatine phosphate stock solution (20 mM final concentration) at 30°C. (The number of repeats can be varied. We did four repeats here).
Stop reaction by adding 450 μl TRIzol at various time points (we stopped at 10, 30, 60, and 90 min reaction time points). RNAs were extracted following the manufacturer’s instructions.
Dissolve the RNA pellet in 10 μl DEPC water. Add 10 μl formamide loading buffer to the solution and boil for 5 min.
Load half of the RNA extraction, as well as 0.5 μl pre-U2.3 substrate (used as input) and 5 μl RNA marker, onto a pre-run 6% denaturing polyacrylamide gel containing 6 M urea.
Run at 120 V in 1× TBE until the loading dye reaches 2/3 of the gel length.
Remove the gel from the electrophoresis apparatus after electrophoresis. Transfer gel onto plastic wrap carefully. Absorb solution with filter paper. Wrap the gel carefully using plastic wrap.
Place the plastic-wrapped gel on the storage phosphor imaging screen and expose for 6-10 h. The phosphor screen is then scanned with the PhosphorImager.
Export the image of the radioactive signal in the ‘tiff’ format (Figure 1).
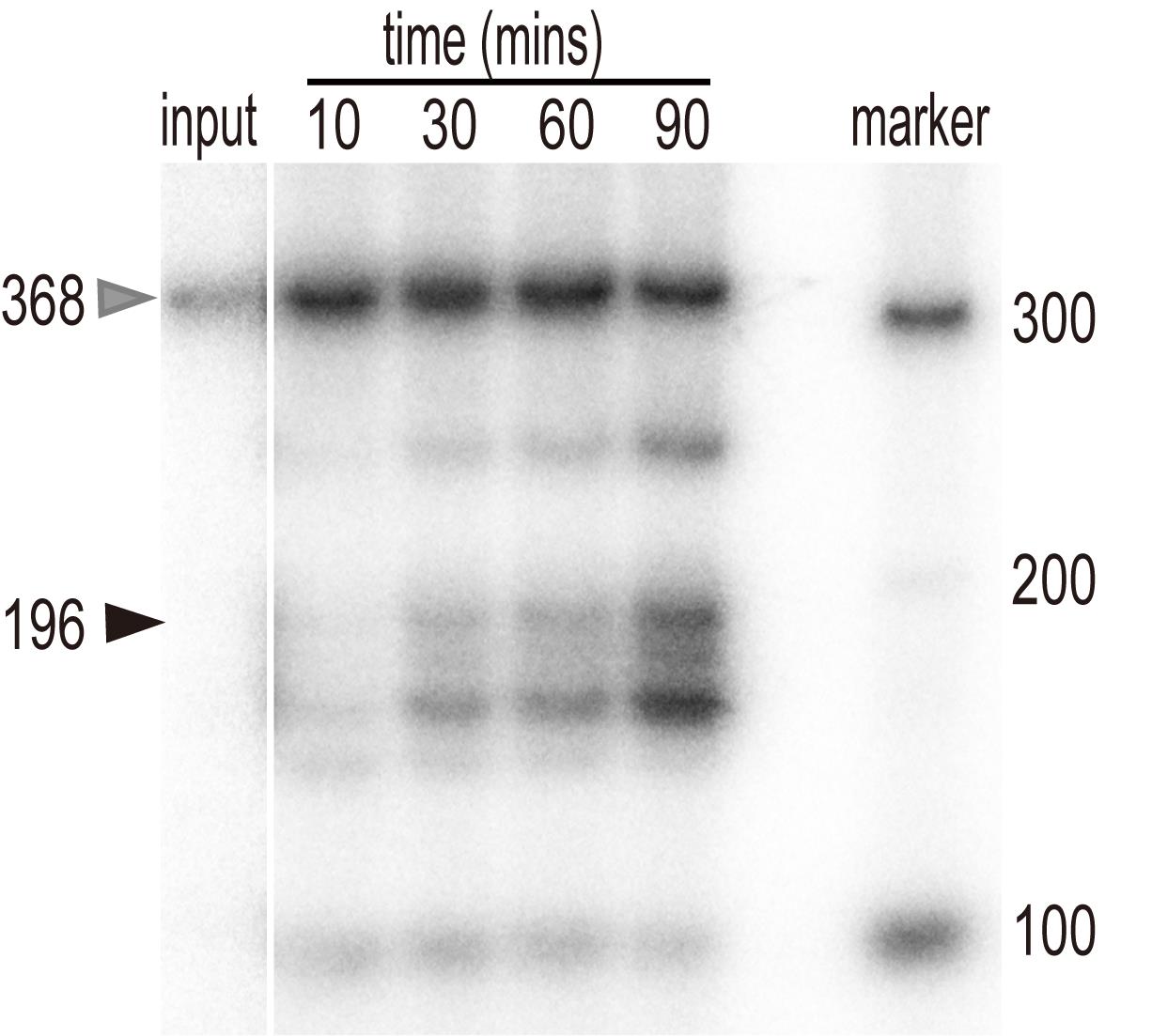
Figure 1. In vitro processing of pre-U2.3-polyG. In vitro transcribed pre-U2.3-polyG RNAs were processed in the nuclear protein extracts from Arabidopsis for various time points, as indicated at the top of the figure. After extraction, RNAs were resolved on a PAGE gel and detected with a PhosphorImager. The position of the pre-U2.3-polyG RNA is indicated by the grey arrow, while the processed mature snRNAs are indicated by the black arrow.
Data analysis
Band intensities were quantified with ImageJ or Quantity One software following these steps (we used Quantity One):
Open the gel image using Quantity One.
Open the ‘Volume Tools’ and select the ‘Volume Rect Tool.’ Select minimum areas of target bands from the image, including the processed mature snRNA and input (Figure 2).
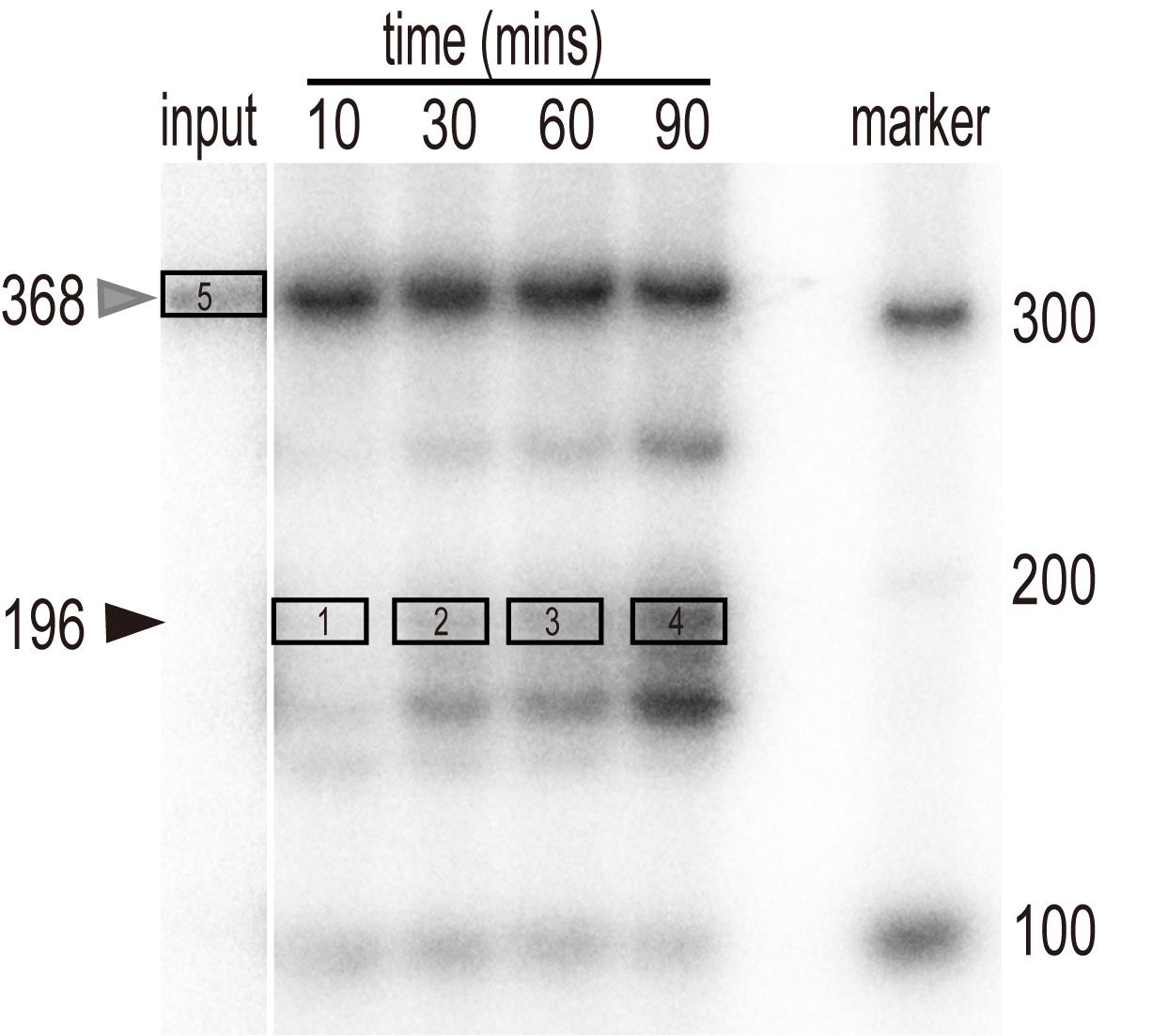
Figure 2. The selected frame of target bands in Quantity One software. Successively select the right processed snRNA bands of various reactions (frame 1-4), according to the relevant position of RNA markers and the pre-U2.3-polyG input (frame 5), using the ‘Volume Rect Tool.’Click the ‘Volume Analysis Report’ and select report options including area name, concentration, mean value, and density (Table 1).
Table 1. Intensity of relevant bands
Use the value of Density to quantify the relative processing efficiency. The intensities of the radioactive signals of various mature U2.3 were normalized to the input.
| Index | Band Name | Volume INT*mm2 | Mean Value INT | Density INT/mm2 |
| 1 | 10 min | 307.75 | 33.13 | 4621.05 |
| 2 | 30 min | 476.26 | 58.59 | 8173.07 |
| 3 | 60 min | 694.97 | 73.95 | 10315.99 |
| 4 | 90 min | 1095.37 | 116.56 | 16259.60 |
| 5 | Input | 829.91 | 72.58 | 10125.62 |
Recipes
Processing buffer
10 mM HEPES, pH 7.9
50 mM KCl
10% glycerol
20 mM creatine phosphate
3 mM MnCl2
2.5% Polyvinyl alcohol
1 mM DTT
Add PMSF, Pepstatin A, and protease inhibitor just before use.
Note: Be sure to make the solution on the day of use.
M1 buffer
10 mM Phosphate buffer, pH 7.8
0.1 M NaCl
10 mM 2-Mercaptoethanol
1 M Hexylene glycol
Add PMSF, Pepstatin A, and protease inhibitor just before use.
M2 buffer
10 mM Phosphate buffer, pH 7.8
0.1 M NaCl
10 mM 2-mercaptoethanol
1 M Hexylene glycol
10 mM MgCl2
Add PMSF, Pepstatin A, and protease inhibitor just before use
M3 buffer
10 mM Phosphate buffer, pH 7.8
0.1 M NaCl
10 mM 2-Mercaptoethanol
Add PMSF, Pepstatin A, and protease inhibitor just before use
2× formamide loading buffer
95% Deionized formamide
0.025% (w/v) Bromophenol blue
0.025% (w/v) Xylene cyanol FF
5 mM EDTA, pH 8.0
6% denaturing polyacrylamide gel containing 6 M urea (10 ml)
2 ml 30% polyacrylamide solution (29:1)
Urea 3.6 g
1ml 5× TBE
9.9 ml water
80 µl 10% APS
10 µl TEMED
Before preparing the gel, clean all components with 70% ethanol and RNase away.
Acknowledgments
This work was supported by grants from the State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources (SKLCUSA-a202008).
Competing interests
The authors declare no competing interests.
References
- Baillat, D., Hakimi, M. A., Naar, A. M., Shilatifard, A., Cooch, N. and Shiekhattar, R. (2005). Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123(2): 265-276.
- Carbon, P., Murgo, S., Ebel, J. P., Krol, A., Tebb, G. and Mattaj, L. W. (1987). A common octamer motif binding protein is involved in the transcription of U6 snRNA by RNA polymerase III and U2 snRNA by RNA polymerase II. Cell 51(1): 71.
- Chen, J. and Wagner, E. J. (2010). snRNA 3' end formation: the dawn of the Integrator complex. Biochem Soc Trans 38(4): 1082-1087.
- Hernandez, N. (2001). Small nuclear RNA genes: a model system to study fundamental mechanisms of transcription. J Biol Chem 276(29): 26733-26736.
- Jawdekar, G. W. and Henry, R. W. (2008). Transcriptional regulation of human small nuclear RNA genes. Biochimica et Biophysica Acta 1779(5): 295-305.
- Liu, Y., Li, S., Chen, Y., Kimberlin, A. N., Cahoon, E. B. and Yu, B. (2016). snRNA 3’ End Processing by a CPSF73-Containing Complex Essential for Development in Arabidopsis. PLoS Biology 14(10): e1002571.
- Vankan, P., Filipowicz, W. (1988). Structure of U2 snRNA genes of Arabidopsis thaliana and their expression in electroporated plant protoplasts. EMBO J 7(3): 791.
- Uguen, P., Murphy, S. (2003). The 3' ends of human pre-snRNAs are produced by RNA polymerase II CTD-dependent RNA processing. EMBO J 22(17): 4544-4554.
- Uguen, P., Murphy, S. (2004). 3'-box-dependent processing of human pre-U1 snRNA requires a combination of RNA and protein co-factors. Nucleic Acids Res 32(10): 2987-2994.
Article Information
Publication history
Accepted: May 17, 2021
Published: Sep 5, 2021
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lin, C., Feng, Y., Peng, X., Wu, J., Wang, W. and Liu, Y. (2021). U2.3 Precursor Small Nuclear RNA in vitro Processing Assay. Bio-protocol 11(17): e4142. DOI: 10.21769/BioProtoc.4142.
Category
Plant Science > Plant molecular biology > RNA
Molecular Biology > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.
![]() Tips for asking effective questions
Tips for asking effective questions
+ Description
Write a detailed description. Include all information that will help others answer your question including experimental processes, conditions, and relevant images.