
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Highly Sensitive Method to Efficiently Profile the Histone Modifications of FFPE Samples
(*contributed equally to this work) Published: Vol 12, Iss 10, May 20, 2022 DOI: 10.21769/BioProtoc.4418 Views: 1730
Reviewed by: Alessandro DidonnaWenfei JiPierre-Damien DenechaudAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics



Related protocols

Abstract
The majority of biopsies in both basic research and translational cancer studies are preserved in the format of archived formalin-fixed paraffin-embedded (FFPE) samples. Profiling histone modifications in archived FFPE tissues is critically important to understand gene regulation in human disease. The required input for current genome-wide histone modification profiling studies from FFPE samples is either 10–20 tissue sections or whole tissue blocks, which prevents better resolved analyses. Nevertheless, it is desirable to consume a minimal amount of FFPE tissue sections in the analysis as clinical tissue of interest are limited. Here, we present FFPE tissue with antibody-guided chromatin tagmentation with sequencing (FACT-seq), highly sensitive method to efficiently profile histone modifications in FFPE tissue by combining a novel fusion protein of hyperactive Tn5 transposase and protein A (T7-pA-Tn5) transposition and T7 in vitro transcription. FACT-seq generates high-quality chromatin profiles from different histone modifications with low number of FFPE nuclei. We showed a very small piece of FFPE tissue section containing ~4000 nuclei is sufficient to decode H3K27ac modifications with FACT-seq. In archived FFPE human colorectal and human glioblastoma cancer tissue, H3K27ac FACT-seq revealed disease specific super enhancers. In summary, FACT-seq allows researchers to decode histone modifications like H3K27ac and H3K27me3 in archival FFPE tissues with high sensitivity, thus allowing us to understand epigenetic regulation.
Graphical abstract:
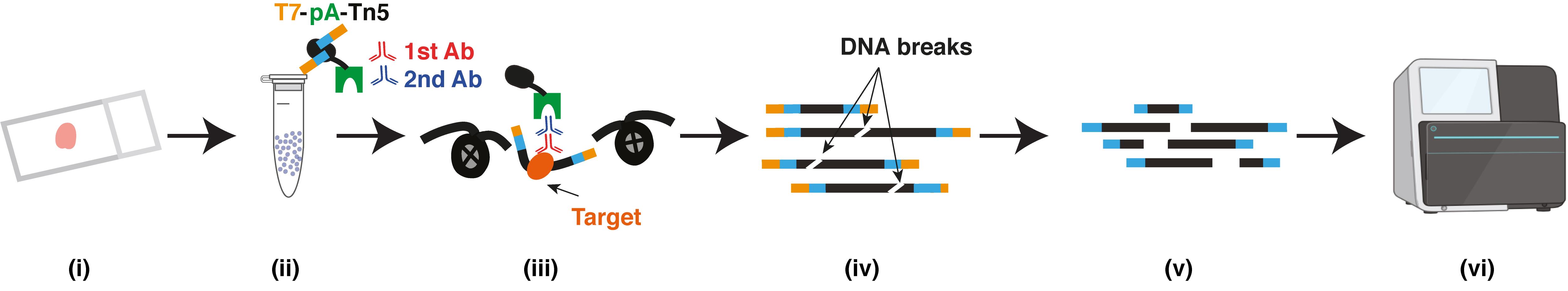
(i) FFPE tissue section; (ii) Isolated nuclei; (iii) Primary antibody, secondary antibody and T7-pA-Tn5 bind to targets; (iv) DNA purification; (v) In vitro transcription and sequencing library preparation; (vi) Sequencing
Background
Currently, Formalin-Fixed Paraffin-Embedding (FFPE) is a universal approach for biopsy specimen processing in basic research and translational studies (Fox et al., 1985; Wang et al., 2005; Haile et al., 2019). Furthermore, it has been reported that large numbers of FFPE specimens are archived worldwide each year (Waldron et al., 2012). More and more techniques have been developed to decipher genomic(Astolfi et al., 2015; Bolognesi et al., 2016), transcriptomic (Pennock et al., 2019), and proteomic (Becker et al., 2017) information in FFPE samples. Nevertheless, a method that can profile epigenetic regulation in FFPE samples with high sensitivity is lacking. Previously, researchers applied chromatin immunoprecipitation with sequencing (ChIP-seq) to archived FFPE tissue. And they have successfully developed pathology tissue chromatin immunoprecipitation (PAT-ChIP) (Fanelli et al., 2010, 2011), fixed-tissue chromatin immunoprecipitation sequencing (FiT-seq) (Cejas et al., 2016), and fixed-tissue ChIP-seq for H3K27 acetylation profiling (FiTAc-seq) (Font-Tello et al., 2020) making it possible to profile histone modifications in FFPE tissue. However, the required input for these technologies from FFPE samples is either 10–20 tissue sections or whole tissue blocks. A higher sample consumption prevents them from being used in a broader application since clinical samples are usually limited. In addition, sonication is included in these available methods, which may potentially introduce sequencing bias (Teytelman et al., 2009). Here, we present a new highly sensitive method, we recently developed, to efficiently profile the histone modifications from FFPE samples using antibody-guided chromatin tagmentation with sequencing (FACT-seq) (Zhao et al., 2021). FACT-seq uses a novel fusion protein (T7-Pa-Tn5) that combines hyperactive Tn5 transposase with protein A, utilizing T7 in vitro transcription to prepare a library, and profile histone modifications in a small amount of FFPE sample.
Materials and Reagents
50 mL Falcon tubes (Sarstedt, catalog number: 62.559.001)
21 G needles (BD Microlance, catalog number: ND432)
27 G needles (BD Microlance, catalog number: 302200)
1 mL syringes (BD Microlance, catalog number: 309628)
1.5 mL Low-bind tubes (Sarstedt, catalog number: 72.706.600)
0.5 mL Qubit tubes (Invitrogen, catalog number: Q32856)
30 μm Filters (Miltenyi Biotech MACS, catalog number: 130-098-458) (room temperature)
Xylene (Histolab, catalog number: 2070) (Room temperature)
Ethanol (VWR BDH Chemicals, catalog number: VWRC20816.552) (room temperature)
Collagenase (Sigma-Aldrich, catalog number: C9263)(-20°C)
Hyaluronidase (Merck Millipore, catalog number: HX0154) (-20°C)
Ampicillin (Serva, catalog number: 69-52-3) (+4°C)
Sodium azide (Merck Millipore, catalog number: 822335) (room temperature)
BSA (Miltenyi Biotech MACS, catalog number: 130-091-376) (+4°C)
IGEPAL CA-630 (Sigma-Aldrich, catalog number: I3021) (+4°C)
1 M CaCl2 (Alfa Aesar, catalog number: J63122) (room temperature)
5 M NaCl (Thermo Fisher Scientific, Invitrogen, catalog number: AM9759) (room temperature)
1 M Tris-HCl pH 8.0 (Thermo Fisher Scientific, Invitrogen, catalog number: 15568-025)
1 M Tris-HCl pH 7.5 (Thermo Fisher Scientific, Invitrogen, catalog number: 15567-027)
1 M MgCl2 (Thermo Fisher Scientific, Invitrogen, catalog number: AM9530G) (room temperature)
FBS (Life Technologies, catalog number: 10108-105) (-20°C)
RNase A (Thermo Fisher Scientific, catalog number: EN0531) (-20°C)
DTT (Thermo Fisher Scientific, catalog number: 20291) (+4°C)
0.5 M EDTA (Thermo Fisher Scientific, Invitrogen, catalog number: AM9260G) (room temperature)
Dimethylformamide (Sigma-Aldrich, catalog number: D4551) (room temperature)
Glycerol (Sigma-Aldrich, catalog number: G9012) (Room temperature)
Sodium deoxycholate (Sigma-Aldrich, catalog number: D6750) (room temperature)
Triton X-100 (Sigma-Aldrich, catalog number: T8787) (room temperature)
Human genomic DNA (Promega, catalog number: G3041) (+4°C)
Qiagen miniElute PCR purification kit (Qiagen, catalog number: 28004)
6× Loading Dye (Thermo Fisher Scientific, catalog number: R0611) (-20°C)
HEPES (Sigma-Aldrich, catalog number: H3375)
Protease inhibitor (Sigma-Aldrich, catalog number: 11873580001)
Spermidine (Sigma-Aldrich, catalog number: S2626)
10% SDS (Thermo Fisher Scientific, Invitrogen, catalog number:1553-035)
Anti-H3K27ac antibody (Abcam, catalog number: ab4729)
Anti-H3K27me3 antibody (Cell Signalling Technology, catalog number: 9733S)
Anti-H3K4me1 antibody (Abcam, catalog number: ab176877)
Anti-H3K36me3 antibody (Abcam, catalog number: ab9050)
Mouse IgG1, kappa monoclonal (Abcam, catalog number: ab18443)
Guinea Pig anti-Rabbit IgG antibody (Antibodies-Online, catalog number: ABIN101961)
Proteinase K (Thermo Fisher Scientific, catalog number: EO0491)
Phenol (Thermo Fisher Scientific, catalog number: 17914)
Chloroform (Sigma-Aldrich, catalog number: C2432)
NEBNext high-fidelity 2× PCR master mix (New England Biolabs, catalog number: M0541S) (-20°C)
SPRIselect beads (Beckman Coulter, catalog number: B23317) (Room temperature)
T7 high yield RNA synthesis kit (New England Biolabs, catalog number: E2040S) (-20°C)
Zymo RNA purification kit (Zymo Research, catalog number: R1013) (room temperature)
SMART MMLV kit (TAKARA, catalog number: 639524) (-20°C)
RNAClean XP beads (Beckman Coulter, catalog number: A63987) (+4°C)
Zymo ChIP DNA clean and concentrator kit (Zymo Research, catalog number: D5205) (room temperature)
40% Acrylamide:bis-acrylamide (Invitrogen, catalog number: HC2040)
10% Ammonium persulfate (Invitrogen, catalog number: HC2005)
TEMED (Invitrogen, catalog number: HC2006)
Digitonin (Millipore, catalog number: 300410)
Nuclease-free water (Invitrogen, catalog number: AM9932)
KOH (Sigma-Aldrich, catalog number: 484016)
50 bp DNA ladder (ThermoFisher Scientific, catalog number: 10488099)
Agilent high sensitive DNA kit (Agilent, catalog number: 5067-4626)
Tn5 transposase [produced in local protein facility following previously description (Picelli et al., 2014)]
pA-Tn5 transposase [produced in local protein facility following previously description (Picelli et al., 2019)]
1× PBS (pH = 7.4) (Thermo Fisher Scientific, catalog number: 10010023)
SYBR gold (ThermoFisher Scientific, catalog number: S33102)
10× TBE Buffer (ThermoFisher Scientific, catalog number: B52)
Mouse kidney/liver FFPE tissue blocks (prepared in the lab)
Buffer used in isolation of single nuclei suspension from FFPE tissue (see Recipes)
Buffers used in Tn5 assembly and activity assay (see Recipes)
Buffers used in epitope retrieval (see Recipes)
Buffers used in antibody binding and tagmentation (see Recipes)
Buffer used in library preparation (see Recipes)
Equipment
Microtome (Leica, Histo Core MULTICUT semi-automated rotary microtome, catalog number:149MULTI0C1, 14051856372 or similar one)
Stereo Microscope (Zeiss stemi DV4 stereo microscope 8x-32x, catalog number: 435421-0000-000 or similar one)
ThermoMixer (Eppendorf, Thermo mixer F1.5, catalog number: 5385000016 or similar one)
Thermal Cycler (Applied Biosystems, Veriti, catalog number: 4375786 or similar thermal cycler)
NanoDrop (Thermo Fisher scientific, Nanodrop 2000c, catalog number: ND2000CLAPTOP or similar one)
Cell Counter (Life Technologies, Countess II, catalog number: AMQAF1000 or similar one)
Centrifuge (Eppendorf centrifuge 5415R, catalog number: EP5415R or similar one)
Rotator (Grant InstrumentsTM 360° Vertical Multi-function Rotator PTR 35, catalog number: 9.721028 or similar one)
Microscopy (Zeiss Imager.Z2, catalog number: 490016-0001-000 or similar one)
DynaMagTM-2 magnetic rack (Thermo Fisher Scientific, catalog number: 12321D or similar one)
Agilent DNA Bioanalyzer (Agilent, catalog number: 5067-4626)
Polyacrylamide gel tray (Thermo Fisher Scientific, catalog number: HC1000S)
Gel documentation system (BIO-RAD, catalog number: 1708195EDU)
Software
Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtmL)
Samtools (http://samtools.sourceforge.net/)
Deeptools (https://deeptools.readthedocs.io/en/develop/content/installation.htmL)
Picard tools (https://broadinstitute.github.io/picard/)
Procedure
Isolation of single nuclei suspension from FFPE block
Sectioning FFPE block into the sections or curls
Deparaffinization and rehydration
Total tissue fragmentation
Disaggregation
Filtration
Tn5 assembly
Tn5 assembly
T7-pA-Tn5 assembly
Tn5 and T7-pA-Tn5 activity assay
Antibody binding and tagmentation
Epitope retrieval
Primary antibody binding
Secondary antibody binding
Tagmentation and reverse cross-linking
DNA isolation and in vitro transcription
DNA purification
Gap filling
SPRI bead purification
In vitro transcription and RNA purification
Reverse transcription and pre-PCR
Reverse transcription
cDNA purification using RNAClean XP beads
Pre-PCR
Second tagmentation and library preparation
Second tagmentation
PCR amplification
Size selection and gel purification
Precise quantification and sequencing
Precise quantification using bioanalyzer
Sequencing
Isolation of single nuclei suspension from FFPE tissue block
FFPE tissues are processed through various steps: deparaffinization, rehydration, total tissue fragmentation, enzymatic digestion, disaggregation, and filtration to isolate single nuclei suspension.
Sectioning the FFPE block into sections (or curls or slides) and preparing the enzymes.
Section the FFPE block at various thickness: 7–20 μm.
Tip: To get good sections without any tissue break, add some ethanol on the top of the tissue block by rubbing with damp ethanol sterile cloth before sectioning it.
Deparaffinization and rehydration
Take one tissue section in a 50 mL Falcon tube, add 1 mL of xylene for deparaffinization, and incubate it for 5 min. After incubation, aspirate the xylene and repeat the same step twice.
After deparaffinization, add 1 mL of 100% ethanol and incubate it for 5 min, followed by sequential rehydration by adding and aspirating different percentages of ethanol (1 mL) (95%, 70%, 50% and 30%). Finally, add 1 mL of nuclease-free water for rehydration and incubate it for 5 min.
Tissue microdissection and enzymatic digestion
After rehydration, the tissue is resuspended in 1 mL of PBS (room temperature) with 0.5 mM of CaCl2, select sections, and use a forceps to transfer tissue to an autoclave glass dish with 200 µL of PBS with 0.5 mM of CaCl2. Perform total tissue fragmentation with two 21 G needles under a stereomicroscope.
Tip: To get suitable nuclei, make tissue as small as possible.
Transfer the fragmented tissue into a 1.5 mL tube with a 1 mL pipette and centrifuge the sample at 3,000 × g (room temperature) for 10 min. After centrifugation, aspirate the supernatant and add 500 µL of 6 mg/mL collagenase (diluted in PBS with 0.5 mM CaCl2), 500 µL of 600 unit/mL hyaluronidase (diluted in PBS with 0.5 mM CaCl2), 50 μg of sodium azide (Merck Millipore, 26628-22-8), 100 μg of ampicillin and incubate for 16 h at 37°C.
Disaggregation
Prepare fresh NST buffer (Nonidet P40 with Salts and Tris buffer) before starting the experiment (see Table 8 for NST buffer recipe). After 16 h of incubation, add 400 μL of NST buffer and centrifuge the mixture at 3,000 × g for 10 min (room temperature), then aspirate and discard the supernatant.
Add 800 μL of NST buffer,10% fetal bovine serum (88 μL) (on ice), and 0.1% DNase-free RNase A (0.88 μL) (on ice) to the pellet and pass through 2 7G needle syringe for at least 30 times for disaggregation.
Filtration
After disaggregation, centrifuge the mixture at 3,000 × g for 10 min, aspirate supernatant, add 800 μL of NST buffer, and filter the mixture twice using a 30 µm filter.
The filtered mixture should be centrifuged at 3,000 × g for 10 min. Then, discard the supernatant first and resuspend the pellet in 500 μL of 1× PBS, take 5 μL of nuclei suspension to count the cells using a cell counter, take 20 μL of nuclei suspension to stain the nuclei with DAPI, and visualize the single nuclei population (Figure 1).
Note: 500,000–1,000 000 nuclei could be gotten from one 20 μm-thick mouse kidney section.
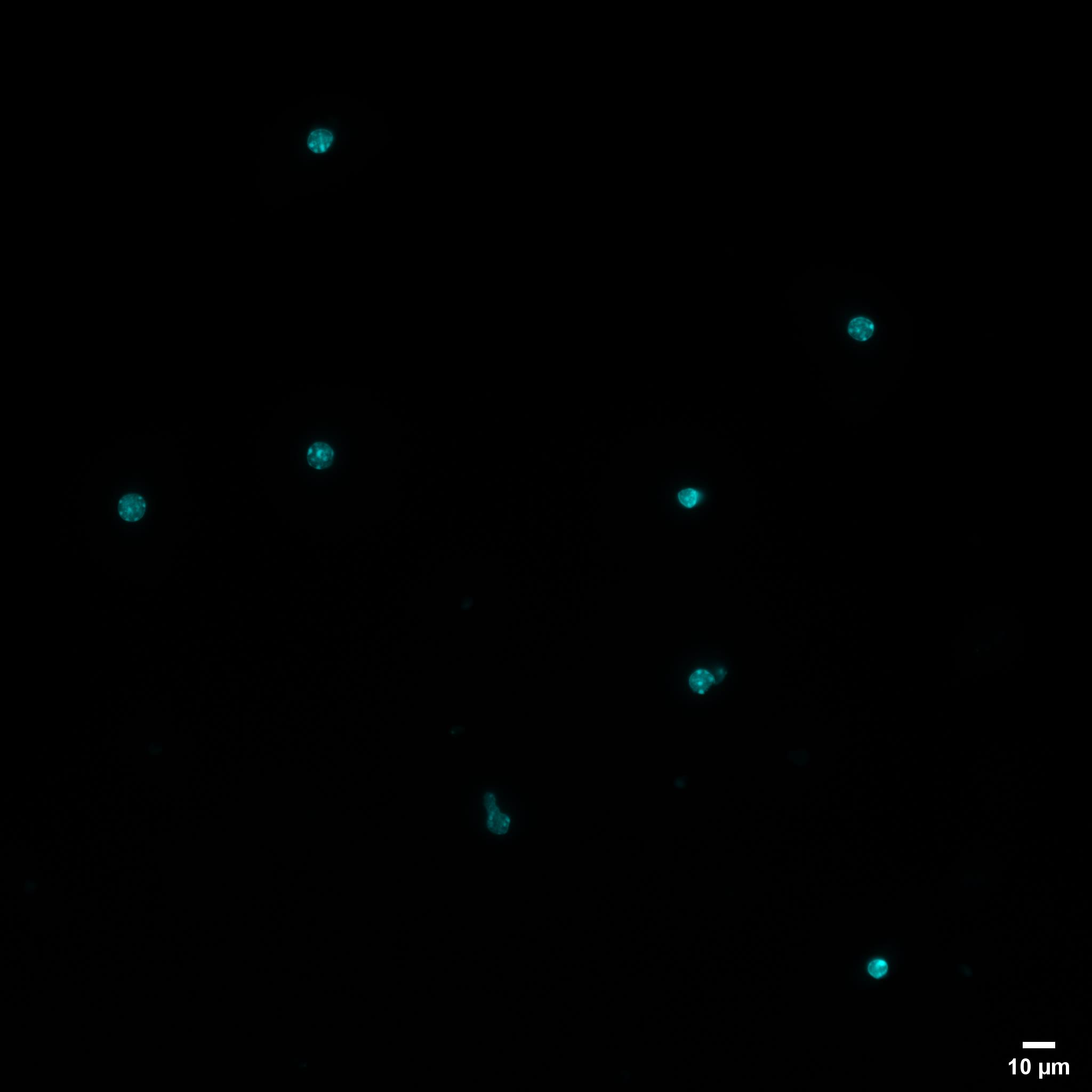
Figure 1. Isolated FFPE mouse nuclei. Representative nuclei isolated from 20-µm-thick mouse FFPE kidney tissue sections and stained with DAPI (Scale bar = 10 µm).
Tn5 assembly
Tn5 assembly mainly covers the steps of loading Tn5 adapter oligos onto Tn5 or pA-Tn5 transposases and checking the Tn5 and T7-pA-Tn5 activity (see Table 1).
Table 1. Summary of assembled Tn5Name Adapters loaded Tn5 Tn5ME-A/Tn5MErev + Tn5ME-B/Tn5MErev T7-pA-Tn5 T7-Tn5ME/Tn5MErev Tip: Prepare 2× dialysis buffer, Tn5, and T7-pA-Tn5 before starting the experiments.
Tn5 Assembly (Table 2)
Tip: The assembled Tn5 will be used in the second tagmentation step.
Table 2. Oligonucleotides for Tn5 assemblyName Oligonucleotides sequence Tn5MErev 5′-[phos]CTGTCTCTTATACACATCT-3′ Tn5ME-A 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′ Tn5ME-B 5′ -GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′ Prepare all DNA oligonucleotides (see Table 1) to 100 μM.
Mix 5 µL of Tn5ME-A with 5 µL of Tn5MErev in one PCR tube, and mix 5 µL of Tn5ME-B with 5 µL of Tn5MErev in another PCR tubes. The DNA sequences of oligonucleotides are list in Table 2.
Perform oligos annealing by subjecting the oligos to 95°C for 5 min followed by slow ramping to 25°C with -0.1°C/s ramping rate using a PCR machine.
After that, mix all the reagents in mentioned volumes: 2 µL of Tn5MErev/Tn5ME-A, 2 µL of Tn5MErev/Tn5ME-B, 20 µL of 100% glycerol, 15.24 2 µL of 2× dialysis buffer (100 mM HEPES−KOH at pH 7.2, 0.2 M NaCl, 0.2 mM EDTA, 2 mM DTT, 0.2% Triton X-100, 20% glycerol), 2.15 µL of Tn5 (46.55 μM), 8.21 µL of water and incubate it at room temperature for 1 h (also see Table 9 for 2× dialysis buffer preparation and Table 10 for the pipetting scheme of Tn5 assembly).
T7-pA-Tn5 assembly (Table 3)
Tip: The assembled T7-pA-Tn5 is used in step C.3.d.
Table 3. Oligonucleotides for T7-pA-Tn5 assemblyName Oligonucleotides sequence Tn5MErev 5′-[phos]CTGTCTCTTATACACATCT-3′ T7-Tn5ME 5′-CATGAGATTAATACGACTCACTATAGGGAGAAGATGTGTATAAGAGACAG-3′ Prepare all oligos (from Table 2) to 100 μM.
Mix the 5 µL of T7-Tn5ME with 5 µL of Tn5MErev in a PCR tube. The DNA sequences of oligonucleotides are list in Table 3.
Perform oligos annealing by subjecting the oligos to 95°C for 5 min followed by slow ramping to 25°C with -0.1°C/s ramping rate using PCR machine.
After that, mix all the reagents in mentioned volumes: 4 µL of T7-Tn5ME/Tn5MErev, 20 µL of 100% glycerol, 15.58 µL of 2× dialysis buffer (100 mM HEPES−KOH at pH 7.2, 0.2 M NaCl, 0.2 mM EDTA, 2 mM DTT, 0.2% Triton X-100, 20% glycerol), 1.81 µL of pA-Tn5 (55.55 μM), 8.61 µL of water and incubate it at room temperature for 1 h (also see Table 11 for the pipetting scheme of T7 pA-Tn5 assembly and Table 8 for 2× dialysis buffer preparation).
Tn5 and T7-pA-Tn5 activity assay
Check the activity of the assembled Tn5, and T7 pA-Tn5 as mentioned in Table 4 (also see Table 12 for 2× TD buffer preparation).
Mix the reagents and incubate the mixture at 55°C for 7 min. After the incubation, purify the mixture with Qiagen MiniElute PCR Purification kit according to the manufacturer protocol and eluted in 10 μL of elution buffer.
Table 4. Mixture of Tn5 or T7-pA-Tn5 activity assayReagents Volume 2× TD buffer 10 μL Human genomic DNA 50 ng Tn5 or T7-pA-Tn5 (2 μM) 1 μL Water Make the final volume to 20 μL/tube Finally, mix with 2 μL of 6× loading dye and run on a 1% agarose gel to check the size of the tagmented DNA along with DNA ladder (Figure 2).
Note: The expected size of fragments of DNA after tagmentation starts from 150 bp to 2,500 bp, and it looks like a smear spreading in the gel lane.
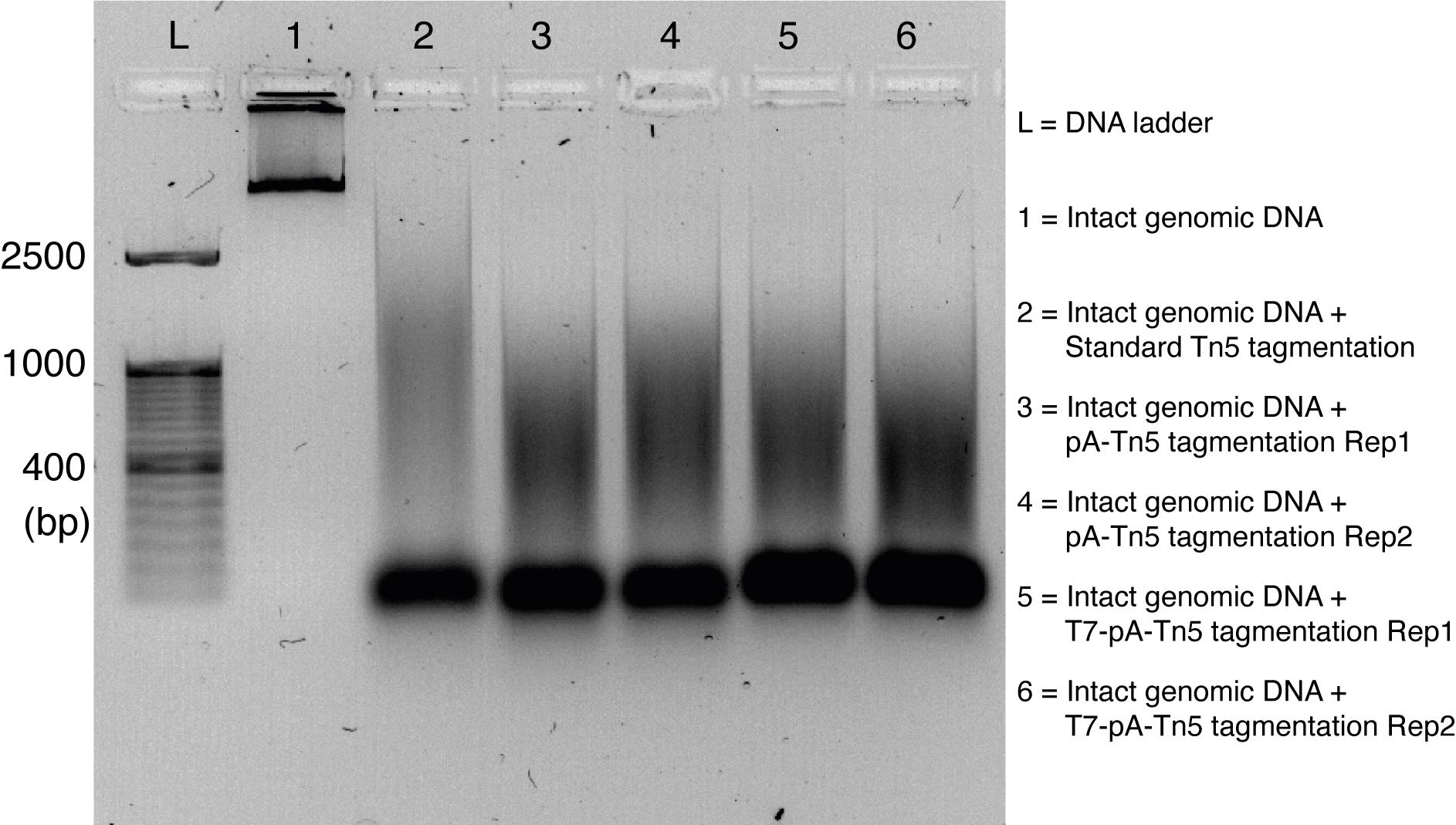
Figure 2. Activity assay of Tn5, pA-Tn5, and T7-pA-Tn5. DNA size distribution after Tn5 transposition showed here. T7-pA-Tn5 has similar activity to Tn5 and pA-Tn5.Antibody binding and tagmentation
Epitope retrieval
After nuclei isolation, transfer a proper amount of FFPE nuclei (1,000–1,500,000) to a new 1.5 mL Lo-Bind tube. Then spin down the nuclei at 3,000 × g for 5 min at room temperature and discard the supernatant.
Note: The number of nuclei used in the experiment depends on the total number of nuclei gotten from nuclei isolation step. The protocol works for 1,000 to 1,500,000 nuclei.
To profile histone modification H3K27ac and H3K4me1 (euchromatin markers), resuspend the FFPE nuclei with 50 μL of Epitope Retrieval Buffer-1 (see below Table 13), and transfer the suspension to a PCR tube. Incubate the nuclei suspension at 50°C for 1 h. To profile histone modification H3K27me3 (heterochromatin marker) and H3K36me3 (gene body marker), a harsher epitope retrieval condition is needed. Therefore, the nuclei are resuspended with 50 μL of Epitope Retrieval Buffer-2 (see below Table 14), and transfer the suspension to a PCR tube. Incubate the nuclei suspension at 65°C for 1 h.
During the incubation time, prepare the FACT-seq Antibody buffer (check more information from the recipe section) and keep it on ice.
After incubation, add 10 μL of 10% Triton X-100 to the tube and transfer the mixture to a 1.5 mL Lo-Bind tube. And place the tube on a shaker, and incubate at 37°C for 30 min with 500 rpm to quench SDS.
After quenching, spin down the mixture at 3,000 × g for 5 min at room temperature and wash the pellet once with the FACT-seq Antibody buffer.
After washing, resuspend the nuclei pellet with 200 μL of FACT-seq Antibody buffer, and use 5 μL of nuclei suspension to count the nuclei number by the cell counter.
Note: Recovery rate after epitope retrieval on average is 62.94%.
Primary antibody binding
After counting nuclei, transfer 100,000 FFPE nuclei into a new 0.5 mL Qubit tube.
Note: Nuclei tend to attach to the tube wall and dry out during the overnight rotation step, and 0.5 mL tubes should be used instead of 1.5 mL tubes.
Centrifuge the nuclei at 2,500 × g for 5 mins at room temperature and discard the supernatant with pipette.
Resuspend the nuclei with at least 1 volume of FACT-seq Antibody buffer.
Centrifuge the nuclei at 2,500 × g for 5 mins at room temperature and discard the supernatant with pipette and resuspend the nuclei with 200 μL of FACT-seq Antibody buffer with 1:100 diluted primary antibody.
Incubated overnight with slow rotation (8–10 rpm) at 4°C.
Tip: Set the rotation speed properly and don’t make the solution stuck to one side of the tube.
Secondary antibody binding
Prepare the following buffers before starting the experiment and keep it on ice or at +4°C.
Tip: We always prepare these buffers freshly.
FACT-seq Dig-washing buffer (see below Table 15)
FACT-seq Dig-300 buffer (see below Table 16)
FACT-seq tagmentation buffer (see below Table 17)
After overnight incubation with primary antibody, spin down the nuclei at 2,000 × g for 5 min at room temperature, wash the nuclei pellet with 200 μL of FACT-seq Dig-washing buffer.
Resuspend the nuclei pellet in 200 μL of FACT-seq Dig-washing buffer with 1:100 diluted secondary antibody (Guinea Pig anti-Rabbit IgG antibody) and incubate the mixture for 1 h at room temperature with slow rotation.
Spin down the mixture at 2,000 × g for 5 min at room temperature, wash it three times with 200 μL of FACT-seq Dig-washing buffer, resuspend the nuclei pellet with 200 μL of FACT-seq Dig-300 buffer containing 1:100 diluted T7 pA-Tn5 (Tip: The T7-pA-Tn5 was assembled in section B.2.). And the protein A domain of T7 pA-Tn5 has the affinity to bind mammalian immunoglobulins, which will bind to the secondary antibody from guinea pig.
Incubated the tubes for 1 h rotating at room temperature.
Tagmentation and reverse crosslinking
After Tn5 binding, spin down the nuclei for 5 min at 600 × g and wash three times with FACT-seq Dig-300 buffer, resuspend the nuclei with 200 μL of FACT-seq tagmentation buffer and incubate the suspension for 1 hr at 37°C.
After that, stop tagmentation by addition of 6.7 μL of 0.5 M EDTA, 22 μL of 10% SDS, and 2.2 μL of 20 mg/mL Proteinase K to each tube, and mix it by gently pipetting up and down several times.
Perform the Proteinase K digestion at 65°C with 1,200 rpm shaking for 2 h.
Perform reverse-crosslinking overnight at 72°C with 1,200 rpm in a heating block for H3K27ac or H3K4me1 (change the overnight temperature to 80°C for H3K27me3 or H3K36me3).
After overnight reverse-crosslinking, add 2.2 µL of 20 mg/mL Proteinase K and perform the Proteinase K digestion again (65°C, 1,200 rpm) for 1 h.
DNA isolation and in vitro transcription
DNA purification
After Proteinase K digestion, purify the mixture with either Qiagen Kit purification or phenol-chloroform purification. (Tip: We don’t see differences in library quality when targeting H3K27ac using these two different purification approaches. When targeting H3K27me3 (heterochromatin marker), phenol-chloroform purification is recommended).
For Qiagen purification, follow the standard protocol of Qiagen MiniElute PCR Purification kit and finally elute the DNA in 20 μL of elution buffer.
For phenol-chloroform purification, after Proteinase K digestion, add elution buffer or sterile water to the tube and make the final volume 300 μL per tube. Add 300 μL of Phenol and mix by full-speed vortexing ~2 s.
Centrifuge at 16,000 × g and 4°C for 15 min. Remove the aqueous layer by pipetting to a fresh 1.5 mL tube and add equal amount of Chloroform.
Invert the tube ~10× to mix, and centrifuge at 16,000 × g and 4°C for 15 min.
Transfer aqueous layer to a fresh 1.5 mL tube, then add 2.5×–3× volume of absolute ethanol and add proper amount of 5M NaCl to make the final concentration become 200 mM (NaCl).
Transfer the tube to -80°C freezer and precipitate the DNA for 2–3 h, and centrifuge the tubes for 20 min 4 °C at 16,000 × g.
Discard the supernatant and add 700 μL of 70% ethanol and slightly vortex it. Centrifuge at 16,000 × g at 4°C for 5 min, then discard the supernatant and air dry the tube for 10–15 min. After drying up, add 25 μL of elution buffer (from Qiagen MiniElution PCR Purification kit) to dissolve the DNA.
Gap filling
Tip: After Tn5 tagmentation, there will be two 9 bp gaps between the mosaic ends and the chromatin strands. And the gap filling is to fill up these two gaps using high-fidelity DNA polymerase.
After DNA purification, transfer the sample to the PCR tube and add an equal volume of high-fidelity 2× PCR master mix.
Then incubate the mixture for 8 mins at 72°C in a thermal cycler.
Purify the sample using the Qiagen MiniElute PCR Purification kit.
SPRI bead purification
Remove the fragments shorter than 150 bp using 1.0× SPRI select beads. Initially, make up the volume up to 50 μL and add 1.0× SPRI, then incubate the mixture for 15 min at room temperature.
After incubation, transfer the tubes to the magnetic rack for 10 min and wash the beads with 150 μL of 80% ethanol twice.
After washing, let the beads dry, add 25 μL of water, incubate it for 10 min at room temperature.
Transfer the tube to the magnetic rack and then carefully transfer the supernatant to a fresh tube.
In vitro transcription and RNA purification
Convert DNA to RNA using T7 RNA synthesis kit with 20 μL reaction system, incubate the mixture at 37 °C for overnight to get a good amount of RNA according to the manufacturer protocol.
Purify the RNA using ZYMO DNase RNA purification kit and finally, elute in 15 μL of elution buffer from the kit. And measure the concentration of RNA using Nanodrop 2000c (see Table 5 for more information).
Tip: We follow the manufactory standard protocol to perform In-vitro transcription and RNA purification. Please refer to the standard protocols from these two kits for detailed procedure.
Table 5. Representative RNA concentration after in-vitro transcription and purification
1The in-vitro transcription was performed for 22–23 h for this particular experimentInitial nuclei number/tube Average RNA concentration1 (ng/μL) Total amount of RNA (μg/tube) A260/280 50000 8898.5 133.5 2.19 25000 8484.2 127.3 2.15 10000 7508.4 112.6 2.11 5000 3215.5 48.2 2.03 2500 1012.7 15.2 1.98 1000 139.0 2.1 1.97
Reverse transcription and pre-PCR
Reverse transcription
Transfer 100 ng RNA to a fresh tube, and perform reverse transcription using TAKARA reverse transcription kit. The final volume of reverse transcription is 22.2 μL/tube.
Tip: We follow the manufactory standard protocol to perform reverse transcription. Please refer to the standard protocol from this kit for detailed procedure.
cDNA purification using RNAClean XP beads
make up the volume up to 40 μL and add 1.8× RNAClean XP beads, incubate the mixture for 15 min at room temperature.
Tip: The “1.8× RNAClean XP beads” mean 1.8 times the volume. 72 μL of RNAClean XP beads is added to every 40 μL reaction.
After incubation, transfer the tubes to the magnetic rack for 10 min and wash the beads with 200 μL of 70% ethanol twice.
After washing, let the beads dry, add 24.2 μL of water, incubate it for 10 min, and transfer the tube to the magnetic rack. Beads bind to the magnetic rack, then carefully transfer the supernatant to a fresh tube.
Pre-PCR
After cDNA purification, convert single-stranded cDNA to double-stranded DNA by adding 25 μL of PCR master mix, 0.8 μL of reverse primer (25 µM) (oligonucleotides sequence is in Table 6).
And then perform pre-PCR (98°C for 10 s,63°C for 30s, 72°C for 1 min, and hold at 10°C, only one cycle) and purify and elute the sample in 24.5 μL, using Qiagen MiniElute PCR Purification kit.
Table 6. Reverse primer sequenceName Oligonucleotides sequence Reverse Primer 5′-CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGGAGATGTG-3′
Second tagmentation and library preparation
Second tagmentation
The second tagmentation aims to add a pair of adapters to the DNA so that the DNA fragments can be amplified and sequenced properly.
Perform tagmentation on the sample by adding 25 μL of 2× TD buffer (20 mM Tris-HCl pH 7.6, 10 mM MgCl2, 20% dimethyl formamide), 0.5 μL of 2 μM Tn5, and incubate at 55°C for 7 min.
After incubation, purify and elute the samples in 24.2 μL of elution buffer, using Qiagen MiniElute PCR Purification kit.
PCR amplification and library preparation
After tagmentation and purification, perform final library amplification by adding 25 μL of 2× PCR master mix, 0.4 μL of forward primer (25 μM), 0.4 μL of reverse primer (25 μM).
Tip: the forward and reverse primers are adapted from the 96 primer pairs in the previous report (Buenrostro et al., 2015). Please refer to the original paper for more information.
Perform PCR amplification (72°C for 5 min, 20 cycles of 98°C for 10 s,63°C for 30 s, 72°C for 1 min and hold at 10°C).
Purify samples with Qiagen MiniElute PCR Purification kit.
Size selection and gel purification
After library amplification and purification, the libraries are run on 8% acrylamide gel.
Mix all of the components in 8% acrylamide gel (see Table 7. for more information) well in a clean 15 mL Falcon tube.
Transfer the mixture to the corresponding polyacrylamide gel tray and let it solidify at room temperature for 30 min.
During the waiting time, prepare DNA ladder and mix all of the samples with 6× DNA loading dye.
Load all the samples and 50 bp DNA ladder on the gel and run at 180 V for 15–20 min.
After running, transfer the gel to 1× SYBR gold and stain the gel in darkness for 20 min.
Table 7. Recipe of 8% acrylamide gel (one gel)Reagents Volume (10 mL) 40% Acrylamide:bis-acrylamide 2 mL 10× TBE buffer 1 mL 10% Ammonium persulfate 50 μL TEMED 10 μL Sterile Water 6.94 mL After staining, check the gel under the gel documentation system and cut each gel lane from 220–1,000 bp by referring to 50 bp DNA ladder.
Transfer each gel cut into a 0.5 mL punched tube, and dip into a 2 mL Eppendorf tube. Then centrifuge both the tubes at 16,000 × g for 5 min and discard the 0.5 mL punched tube.Note: The aim of this step is to make the entire gel lane into small pieces.
Add 300 μL of crush soak buffer (500 mM NaCl, 1 mM EDTA, 0.5% SDS [Table 18]) to the gel and incubate the mixture at 55°C for 8 h at least to dissolve DNA fragments in the buffer.Note: The aim of this step is to dissolve DNA fragments in the buffer.
After incubation, transfer the gel mixture to costar tubes and centrifuge for 5 min at 16,000 × g.
Then collect the solution passing through the costar filter and use Zymo ChIP DNA Clean and concentrator kit to purify it and finally elute in 15 μL.
Tip: We follow the manufactory standard protocol to perform DNA purification. Please refer to the standard protocol from this kit for detailed procedure.
Precise quantification and sequencing
Precise quantification DNA concentration of FACT-seq libraries using Agilent high sensitivity DNA kit following manufacturing protocol.
Tip: Please refer to the protocol from this kit for detailed procedure.Sequencing: Sequence the FACT-seq libraries on Illumina NovaSeq 6000 sequencer or Illumina MiniSeq platform with paired end sequencing, and some results (Zhao et al., 2021) are as shown in Figure 3.
Tip: Please refer to the protocol from Illumina for detailed procedure.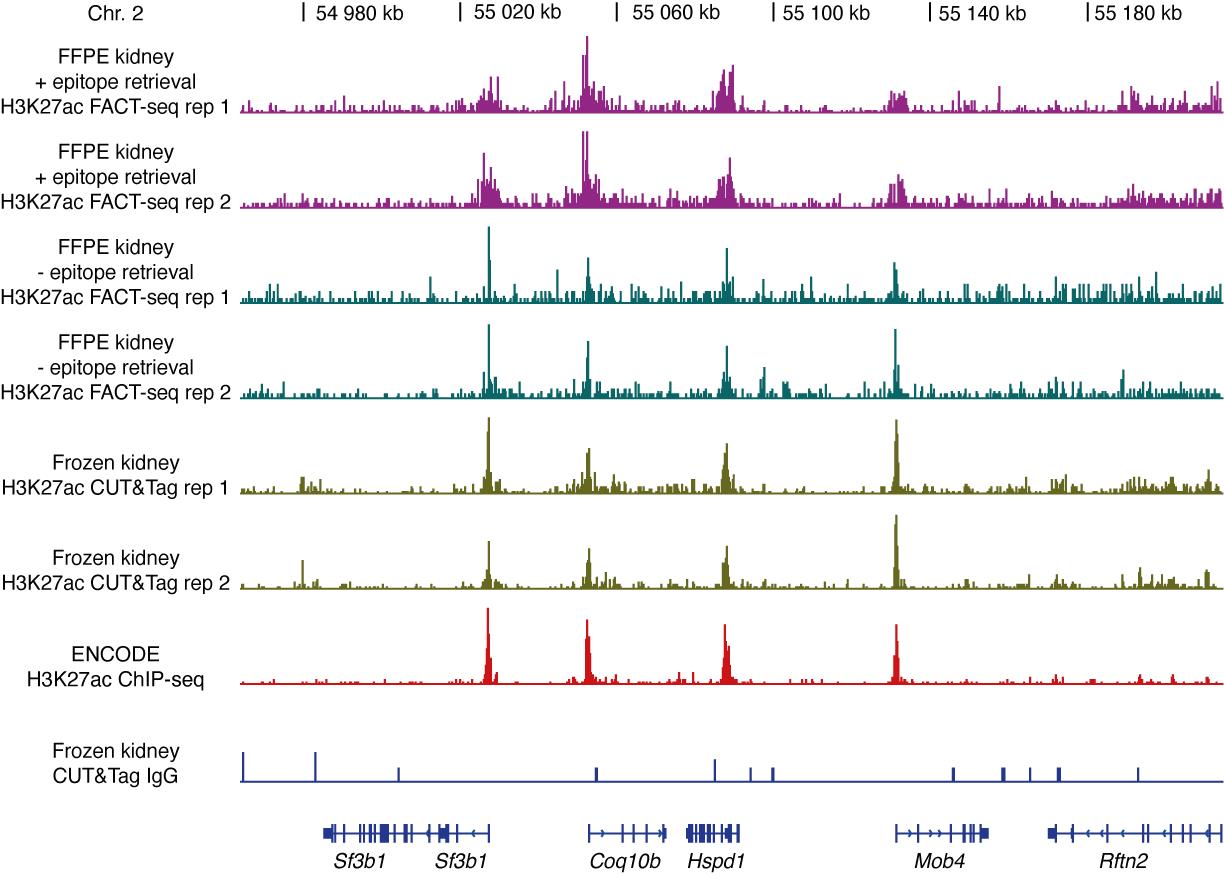
Figure 3. Representative genome browser tracks of H3K27ac sequencing libraries from mouse kidney nuclei, adopted from Zhao et al. (2021). Results from H3K27ac FACT-seq of mouse FFPE kidney nuclei (- epitope retrieval), H3K27ac FACT-seq of mouse FFPE kidney nuclei (+ epitope retrieval), H3K27ac CUT&Tag of frozen mouse kidney nuclei, ENCODE H3K27ac ChIP-seq of frozen mouse kidney nuclei, and CUT&Tag IgG control of mouse kidney nuclei. The gene names are shown at the bottom. Chr = Chromosome. For in detail analysis, refer to FACT-seq paper (Zhao et al., 2021).
Data analysis
Only reads one (R1) will be involved in the downstream analysis. We first adopt levenshtein distance algorithm to map T7 promoter sequence to each read in the adaptor trimming procedure; then T7 promoter sequences could be trimmed from each read in the fastq file with in-house script custom script (https://github.com/pengweixing/FACT).
- The trimmed fastq file of human samples and mouse samples are mapped to the hg19/hg38 reference genome or mm9/mm10 reference genome respectively using bowtie2 v.2.3.5 (Langmead and Salzberg, 2012) with the parameters --end-to-end --very-sensitive -I 10 -X 700. The aligned BAM files are sorted by samtools v.1.9 (Li et al., 2009). The duplicate reads are removed with Picard v1.79 (http://picard.sourceforge.net), and be filtered with alignment quality of >q2. For the peak calling, broad peaks could be called using SICER (Zhang et al., 2008) with the following parameters gap size = 600 bp and window size = 200 bp. Narrow peaks could be called using SICER with following parameters gap size = 400 bp and window size = 200 bp. The bigwig files were generated from BAM file using deeptools (Ramirez et al., 2014) with the parameters bamCoverage -normalizeUsing CPM. The peaks of sequencing libraries are visualized by IGV (Thorvaldsdottir et al., 2013) software.
Notes
- Be extra careful when working with detergents like sodium deoxycholate, digitonin, and SDS. Remember to wear a face mask, lab coat, and gloves when dissolving the powder of sodium deoxycholate and digitonin in the liquid.
- We have successfully profiled H3K27ac in FFPE mouse nuclei with a limited amount of input (1,000 nuclei/tube or starting nuclei isolation from a small piece of tissue containing approximately 5,000 nuclei). So, the FACT-seq protocol could usually work even if the nuclei input is less than 100,000 nuclei/tube.
- Epitope retrieval condition is an important steps to obtain a good library quality. Here, we provided the best epitope retrieval conditions for H3K27ac, H3K27me3, H3K4me1, and H3K36me3. This epitope retrieval condition may directly be used for the other histone modifications, including H3K4me3, H3K9me2, or H3K9me3.
- FACT-seq targeting H3K27ac could be a positive control if you want to test unknown conditions.
- The DNA amount we got after tagmentation and purification range from 100 ng–300 ng/tube (each tube starts from 100,000 FFPE nuclei). We would always proceed with library preparation unless the DNA concentration is too low (< 10 ng/tube) or the purity of the DNA is too bad (A260/280 < 1.5).
- We have successfully performed FACT-seq in FFPE human colorectal cancer and FFPE human glioblastoma samples. The RNA yield after in-vitro transcription varies when using different samples or profiling different markers. If the RNA yield of a particular sample is low, you could try extending the time in-vitro transcription from overnight (8 h) to 16 h or even longer. The longest in-vitro transcription we performed was approximately 23 h, and we still got good libraries.
- For RNA purification, we initially used the traditional TRIzol RNA purification and got good results. Since TRIzol purification is time-consuming, we finally switched to using the kit purification.
Recipes
- Buffer used in isolation of single nuclei suspension from FFPE tissue section
Table 8. Recipe of NST buffer (Nonidet P40 with Salts and Tris buffer)Reagents Initial Concentration Final Concentration Dilution Fold Final Volume (10 mL) NaCl 5,000 mM 146 mM 34.25 292 μL Tris-HCl pH 7.5 1,000 mM 5 mM 200 50 μL Tris-HCl pH 8 1,000 mM 5 mM 200 50 μL CaCl2 1,000 mM 1 mM 1000 10 μL MgCl2 1,000 mM 21 mM 47.62 210 μL BSA 10% 0.05% 200 50 μL IGEPAL CA-630 10% 0.20% 50 200 μL Nuclease-free water / / / 9138 μL
Collagenase: 6 mg/mL in PBS with 0.5 mM CaCl2
Hyaluronidase: 600 units/mL in PBS with 0.5 mM CaCl2 - Buffers used in Tn5 assembly and activity assay
Table 9. Recipe of 2× Dialysis bufferReagents Initial Concentration Final Concentration Dilution Fold Final Volume (10 mL) HEPES(K+) pH 7.2 500 mM 100 mM 5 2 mL NaCl 5,000 mM 200 mM 25 400 μL EDTA 500 mM 0.2 mM 2500 4 μL DTT 500 mM 2 mM 250 40 μL Triton X-100 10% 0.20% 50 200 μL Glycerol 100% 20% 5 2 mL Nuclease-free water / / / 5,356 μL
Table 10. Recipe of Tn5 assemblyReagents Initial Concentration Final Concentration Dilution Fold Final Volume (50 μL) Tn5MErev/Tn5ME-A 50 μM 2 μM 25 2 μL Tn5MErev/Tn5ME-B 50 μM 2 μM 25 2 μL Glycerol 100% 40% 2.5 20 μL 2× Dialysis buffer / / / 15.24 μL Tn5 46.55 μM 2 μM 23.26 2.15 μL Nuclease-free water / / / 8.61 μL
Table 11. Recipe of T7 pA-Tn5 assemblyReagents Initial Concentration Final Concentration Dilution Fold Final Volume (50 μL) Tn5MErev/Tn5ME-A 50 μM 4 μM 12.5 4 μL Glycerol 100% 40% 2.5 20 μL 2× Dialysis buffer / / / 15.58 μL pA-Tn5 55.55 μM 2 μM 27.76 1.81 μL Nuclease-free water / / / 8.61 μL
Table 12. Recipe of 2× TD bufferReagents Initial Concentration Final Concentration Dilution Fold Final Volume (10mL) Tris-HCl pH 7.5 1,000 mM 20 mM 50 200 μL MgCl2 1,000 mM 10 mM 100 100 μL Dimethyl formamide 100% 20% 5 2 mL Nuclease-free water / / / Bring up to 10 mL - Buffers used in epitope retrieval
Table 13. Recipe of Epitope Retrieval Buffer-1Reagents Initial Concentration Final Concentration Dilution Fold Final Volume (10 mL) EDTA 500 mM 10 mM 50 200 μL Tris-HCl pH 8 1,000 mM 50 mM 20 500 μL SDS 10% 0.10% 100 100 μL Nuclease-free water / / / 9.2 mL
Table 14. Recipe of Epitope Retrieval Buffer-2Reagents Initial Concentration Final Concentration Dilution Fold Final Volume (10 mL) EDTA 500 mM 10 mM 50 200 μL Tris-HCl pH 8 1,000 mM 50 mM 20 500 μL SDS 10% 0.10% 100 100 μL Sodium deoxycholate 10% 0.10% 100 100 μL Nuclease-free
water/ / / 9.1 mL - Buffers used in antibody binding and tagmentation
Table 15. Recipe of FACT-seq Dig-washing buffer (for 8 libraries)Reagents Initial Concentration Final Concentration Dilution Fold Final Volume (15 mL) HEPES(K+) pH 7.6 500 mM 20 mM 25 600 μL NaCl 5,000 mM 150 mM 33.33 450 μL Spermidine 2,000 mM 0.5 mM 4,000 3.75 μL Digitonin 5% 0.05% 100 150 μL IGEPAL CA-630 10% 0.01% 1,000 15 μL BSA 10% 1% 10 1,500 μL Nuclease-free water
(with protease inhibitor added)/ / 12.28 mL
To prepare FACT-seq Antibody buffer (for 8 libraries), transfer 4 mL of FACT-seq Dig-washing buffer to a new tube and add 16 μL of 0.5 M EDTA, mix well and keep it on ice.
Table 16. Recipe of FACT-seq Dig-300 buffer (for 8 libraries)Reagents Initial Concentration Final Concentration Dilution Fold Final Volume (10 mL) HEPES(K+) pH 7.6 500 mM 20 mM 25 400 μL NaCl 5,000 mM 300 mM 16.67 600 μL Spermidine 2,000 mM 0.5 mM 4,000 2.5 μL Digitonin 5% 0.05% 100 100 μL IGEPAL CA-630 10% 0.01% 1,000 100 μL BSA 10% 1% 10 1,000 μL Nuclease-free water
(with protease inhibitor added)/ / 7.89 mL
Table 17. Recipe of FACT-seq tagmentation buffer (for 8 libraries)Reagents Initial Concentration Final Concentration Dilution Fold Final Volume (2 mL) HEPES (K+) pH 7.6 500 mM 20 mM 25 80 μL NaCl 5,000 mM 300 mM 16.67 120 μL Spermidine 2,000 mM 0.5 mM 4,000 0.5 μL Digitonin 5% 0.05% 100 20 μL IGEPAL CA-630 10% 0.01% 1000 2 μL MgCl2 1,000 mM 10 mM 100 20 μL Nuclease-free water
(with protease inhibitor added)/ / 1,757.5 μL - Buffer used in library preparation
Table 18. Recipe of crush soak bufferReagents Initial Concentration Final Concentration Dilution Fold Final Volume (10 mL) NaCl 5,000 mM 500 mM 10 1,000 μL EDTA 500 mM 1 mM 500 20 μL SDS 10% 0.50% 20 500 μL Nuclease-free water / / / 8,480 μL
Acknowledgments
This protocol is derived from the previous publication FACT-seq from Chen Lab (Zhao et al., 2021). We want to thank the contributions of all of Chen Lab’s members who involved in this work. We want to thank Protein Science Facility at Karolinska Institute for providing purified Tn5 and pA-Tn5. Our funding sources including: Swedish Research Council [VR-2016-06794, VR-201702074 to X.C.]; Åke Wibergs stiftelse [M20-0007 to X.C.];Beijer Foundation (to X.C.); Jeassons Foundation (to X.C.); Petrus och Augusta Hedlunds Stiftelse (to X.C.); Göran Gustafsson’s prize for younger researchers (to X.C.); Vleugel Foundation (to X.C.); Linnéstiftelsen for medicinsk forskning (to X.C.); Uppsala University (to X.C.); Swedish Cancer Society (CAN 2021-1449Pj, 22 0491 JIA to X.C.).
Competing interests
Xingqi Chen, Vamsi Krishna Polavarapu, and Linxuan Zhao have filed patent applications related to previous published FACT-seq paper and the protocol described here. The title of the patent application is “Method of preparing DNA from formalin-fixed-paraffin-embedded (FFPE) tissue samples”. The Swedish Provisional Application was filed on 28 June 2021, Patent Application No. 2150823-9 in Sweden. The authors declare no competing financial interests.
References
- Astolfi, A., Urbini, M., Indio, V., Nannini, M., Genovese, C. G., Santini, D., Saponara, M., Mandrioli, A., Ercolani, G., Brandi, G., et al. (2015). Whole exome sequencing (WES) on formalin-fixed, paraffin-embedded (FFPE) tumor tissue in gastrointestinal stromal tumors (GIST). BMC Genomics 16: 892.
- Becker, J. S., McCarthy, R. L., Sidoli, S., Donahue, G., Kaeding, K. E., He, Z., Lin, S., Garcia, B. A. and Zaret, K. S. (2017). Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol Cell 68(6): 1023-1037 e1015.
- Bolognesi, C., Forcato, C., Buson, G., Fontana, F., Mangano, C., Doffini, A., Sero, V., Lanzellotto, R., Signorini, G., Calanca, A., et al. (2016). Digital Sorting of Pure Cell Populations Enables Unambiguous Genetic Analysis of Heterogeneous Formalin-Fixed Paraffin-Embedded Tumors by Next Generation Sequencing. Sci Rep 6: 20944.
- Buenrostro, J. D., Wu, B., Litzenburger, U. M., Ruff, D., Gonzales, M. L., Snyder, M. P., Chang, H. Y. and Greenleaf, W. J. (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523(7561): 486-490.
- Cejas, P., Li, L., O'Neill, N. K., Duarte, M., Rao, P., Bowden, M., Zhou, C. W., Mendiola, M., Burgos, E., Feliu, J., et al. (2016). Chromatin immunoprecipitation from fixed clinical tissues reveals tumor-specific enhancer profiles. Nat Med 22(6): 685-691.
- Fanelli, M., Amatori, S., Barozzi, I. and Minucci, S. (2011). Chromatin immunoprecipitation and high-throughput sequencing from paraffin-embedded pathology tissue. Nat Protoc 6(12): 1905-1919.
- Fanelli, M., Amatori, S., Barozzi, I., Soncini, M., Dal Zuffo, R., Bucci, G., Capra, M., Quarto, M., Dellino, G. I., Mercurio, C., et al. (2010). Pathology tissue-chromatin immunoprecipitation, coupled with high-throughput sequencing, allows the epigenetic profiling of patient samples. Proc Natl Acad Sci U S A 107(50): 21535-21540.
- Font-Tello, A., Kesten, N., Xie, Y., Taing, L., Vareslija, D., Young, L. S., Hamid, A. A., Van Allen, E. M., Sweeney, C. J., Gjini, E., et al. (2020). FiTAc-seq: fixed-tissue ChIP-seq for H3K27ac profiling and super-enhancer analysis of FFPE tissues. Nat Protoc 15(8): 2503-2518.
- Fox, C. H., Johnson, F. .B, Whiting, J. and Roller, P. P. (1985). Formaldehyde fixation. J Histochem Cytochem 33(8): 845-53.
- Haile, S., Corbett, R. D., Bilobram, S., Bye, M. H., Kirk, H., Pandoh, P., Trinh, E., MacLeod, T., McDonald, H., Bala, M., et al. (2019). Sources of erroneous sequences and artifact chimeric reads in next generation sequencing of genomic DNA from formalin-fixed paraffin-embedded samples. Nucleic Acids Res 47(2): e12.
- Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., Ahmad, K. and Henikoff, S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10(1): 1930.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Pennock, N. D., Jindal, S., Horton, W., Sun, D., Narasimhan, J., Carbone, L., Fei, S. S., Searles, R., Harrington, C. A., Burchard, J., et al. (2019). RNA-seq from archival FFPE breast cancer samples: molecular pathway fidelity and novel discovery. BMC Med Genomics 12(1): 195.
- Picelli, S., Bjorklund, A. K., Reinius, B., Sagasser, S., Winberg, G. and Sandberg, R. (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24(12): 2033-2040.
- Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. and Manke, T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42(Web Server issue): W187-191.
- Teytelman, L., Ozaydin, B., Zill, O., Lefrancois, P., Snyder, M., Rine, J. and Eisen, M. B. (2009). Impact of chromatin structures on DNA processing for genomic analyses. PLoS One 4(8): e6700.
- Thorvaldsdottir, H., Robinson, J. T. and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2): 178-192.
- Waldron, L., Simpson, P., Parmigiani, G. and Huttenhower, C. (2012). Report on emerging technologies for translational bioinformatics: a symposium on gene expression profiling for archival tissues. BMC Cancer 12: 124.
- Wang, Y., Moorhead, M., Karlin-Neumann, G., Falkowski, M., Chen, C., Siddiqui, F., Davis, R. W., Willis, T. D. and Faham, M. (2005). Allele quantification using molecular inversion probes (MIP). Nucleic Acids Res 33(21): e183.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W., et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9(9): R137.
- Zhao, L., Xing, P., Polavarapu, V. K., Zhao, M., Valero-Martinez, B., Dang, Y., Maturi, N., Mathot, L., Neves, I., Yildirim, I., et al. (2021). FACT-seq: profiling histone modifications in formalin-fixed paraffin-embedded samples with low cell numbers. Nucleic Acids Res 49(21): e125.
Article Information
Publication history
Accepted: Mar 30, 2022
Published: May 20, 2022
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhao, L., Polavarapu, V. K., Yadav, R. P., Xing, P. and Chen, X. (2022). A Highly Sensitive Method to Efficiently Profile the Histone Modifications of FFPE Samples. Bio-protocol 12(10): e4418. DOI: 10.21769/BioProtoc.4418.
Category
Cancer Biology > General technique > Molecular biology technique
Medicine
Molecular Biology > DNA > Chromatin accessibility
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.
![]() Tips for asking effective questions
Tips for asking effective questions
+ Description
Write a detailed description. Include all information that will help others answer your question including experimental processes, conditions, and relevant images.