
- Submit a Protocol
- Receive Our Alerts
- EN

- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Gene Replacement by a Selectable Marker in the Filamentous Fungus Magnaporthe oryzae
Published: Vol 13, Iss 17, Sep 5, 2023 DOI: 10.21769/BioProtoc.4809 Views: 292
Reviewed by: Amey RedkarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Magnaporthe oryzae is a filamentous fungus responsible for the detrimental rice blast disease afflicting rice crops worldwide. For years, M. oryzae has served as an excellent model organism to study plant pathogen interactions due to its sequenced genome, its amenability to functional genetics, and its capacity to be tracked in laboratory settings. As such, techniques to genetically manipulate M. oryzae for gene deletion range from genome editing via CRISPR-Cas9 to gene replacement through homologous recombination. This protocol focuses on detailing how to perform gene replacement in the model organism, M. oryzae, through a split marker method. This technique relies on replacing the open reading frame of a gene of interest with a gene conferring resistance to a specific selectable chemical, disrupting the transcription of the gene of interest and generating a knockout mutant M. oryzae strain.
Key features
• Comprehensive overview of primer design, PEG-mediated protoplast transformation, and fungal DNA extraction for screening.
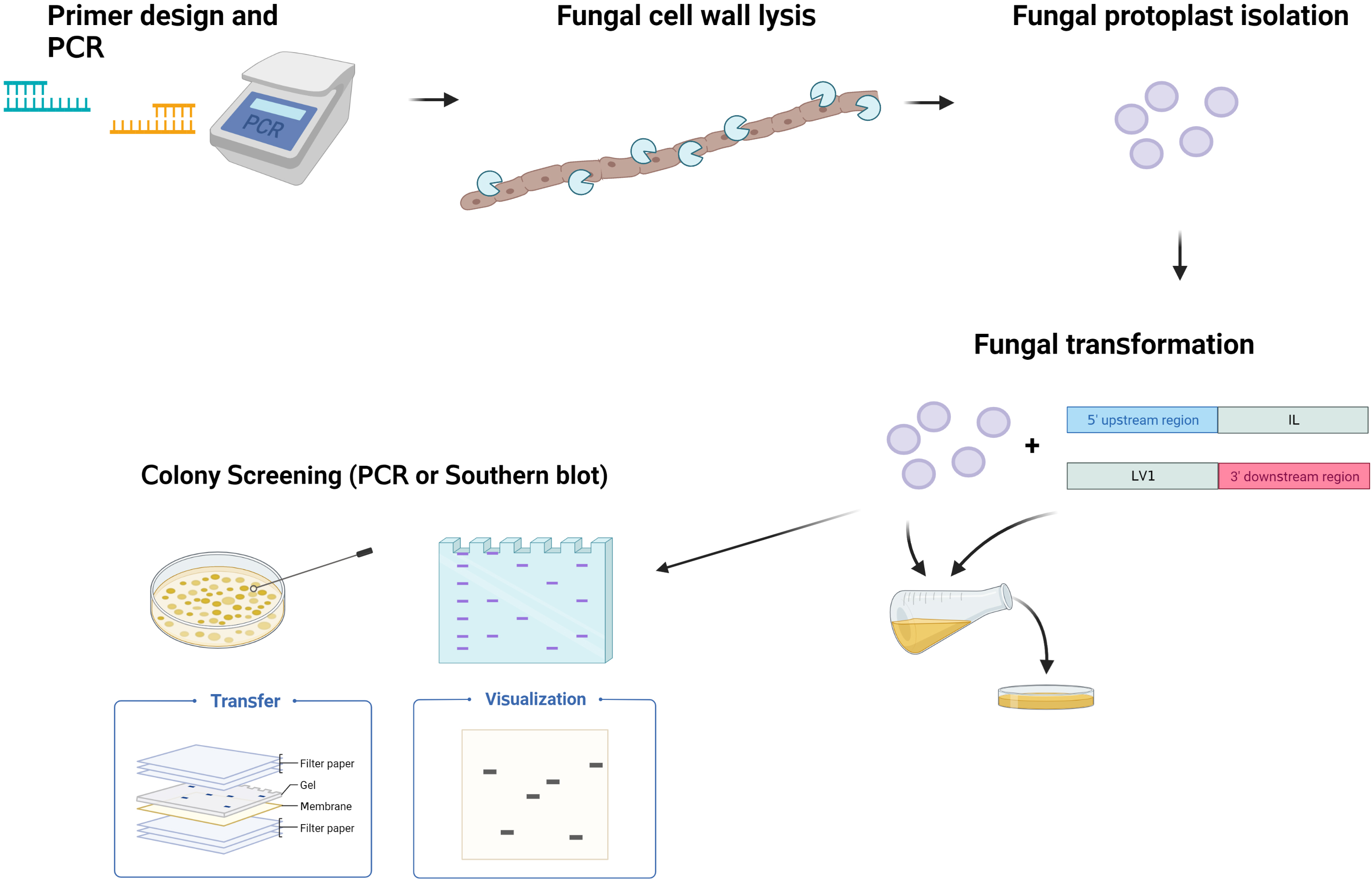
Graphical overview
Background
The fungal plant pathogen Magnaporthe oryzae is the causal agent of rice blast disease. Rice blast afflicts all parts of rice, including leaves, necks, and panicles. Due to its severity, rice blast continues to annihilate rice crops worldwide every year (Fernandez and Orth, 2018; Asibi et al., 2019). As such, dissecting the intricacies of M. oryzae biology is vital to further our understanding of this devastating plant pathogen. As an organism that can be cultivated in the lab as well as genetically manipulated, M. oryzae serves as a model organism to elucidate the molecular basis of rice blast disease and plant pathogen interactions. M. oryzae genetic transformations enable researchers to characterize protein functionality through gene deletions or visualize protein localization through fluorescent tagging. Through editing of the M. oryzae genome, methods to prevent the spread of this disease may be developed. For years, the split marker method has been a key approach for gene deletions in M. oryzae (Sweigard et al., 1995; Fernandez et al., 2014a, 2014b, and 2021). Relying on the homologous recombination that occurs naturally in M. oryzae, the split marker method entails generating flanking regions of a gene of interest through two rounds of PCR and fusing each of them to half of a selectable marker, without requiring any subcloning. This method relies on three homologous recombination events occurring, with two events fusing the flank regions to each half of the selectable marker, and one fusing the selectable marker cassette together. Combining these flanks with M. oryzae protoplasts will generate mutants that have replaced the gene of interest with the selectable marker. In more recent times, various methods for gene editing have been reported. For instance, CRISPR-Cas9-based approaches have been applied for gene editing in M. oryzae (Foster et al., 2018). However, the split marker method has been widely utilized throughout the years due to its convenient, effective, and inexpensive features that facilitate the generation of mutant strains in M. oryzae.
Materials and reagents
Biological materials
Magnaporthe oryzae entry strain of choice, such as Guy11 as used in this protocol (Guy11 strain can be obtained from the Fungal Genetics Stock Center).
Reagents
Yeast nitrogen base w/o amino acids (BD Difco, catalog number: 233520)
Ammonium nitrate 98%, ACS reagent (Thermo Fisher, catalog number: 423350250)
L-Asparagine, 99% (Thermo Fisher, catalog number: B21473.36)
Dextrose/D-(+)-glucose anhydrous ACS reagent grade (MP Biomedicals, catalog number: 152527)
Sucrose (Thermo Fisher, catalog number: J65148.36)
Agar (Sigma-Aldrich, catalog number: A1296-100G)
Peptone (Thermo Fisher, catalog number: 211677)
Yeast extract (Thermo Fisher, catalog number: 212750)
Casamino acids (Thermo Fisher, catalog number: 228820)
NaOH (Sigma-Aldrich, catalog number: S5881-500G)
Na2HPO4 (Sigma-Aldrich, catalog number: S9763-100G)
100% ethanol (Thermo Fisher, catalog number: T038181000)
100% isopropanol (Thermo Fisher, catalog number: T036181000)
RNase (Sigma-Aldrich, catalog number: 10109142001)
Sodium nitrate (Sigma-Aldrich, catalog number: 221341-500G)
Potassium chloride (Thermo Fisher, catalog number: A11662.0B)
Magnesium sulfate heptahydrate (Thermo Fisher, catalog number: A14491.0B)
Potassium phosphate, dibasic (Sigma-Aldrich, catalog number: P3786-100G)
Zinc sulphate heptahydrate (Sigma-Aldrich, catalog number: Z0635-100G)
Boric acid (VWR, catalog number: BDH9222-500G)
Manganese chloride (Thermo Fisher, catalog number: 271410250)
Iron sulfate heptahydrate, ACS, 99+% (Alta Aesar, catalog number: 14498-30)
Cobalt chloride hexahydrate (Thermo Fisher, catalog number: 011344.30)
Copper sulfate pentahydrate (Thermo Fisher, catalog number: 197722500)
Sodium molybdate (Thermo Fisher, catalog number: 206371000)
Na4EDTA (Thermo Fisher, catalog number: J15700.A1)
Biotin (Sigma-Aldrich, catalog number: B4501-100MG)
Pyridoxin (Sigma-Aldrich, catalog number: P5669-25G)
Thiamine (Thermo Fisher, catalog number: 148991000)
Riboflavin (Sigma-Aldrich, catalog number: 47861)
PABA (Sigma-Aldrich, catalog number: A9878-5G)
Nicotinic acid (Sigma-Aldrich, catalog number: N0761-100G)
1 M Tris-HCl, pH 8.0 (Thermo Fisher, catalog number: 15568025)
0.5 M EDTA, pH 8.0 (Sigma-Aldrich, catalog number: 324506-100ML)
Sorbitol (Thermo Fisher, catalog number: 036404.A3)
Lysing enzymes from Trichoderma (Sigma-Aldrich, catalog number: L1412)
1 M Tris-HCl, pH 7.5 (Thermo Fisher, catalog number: 15567027)
1 M CaCl2 (Thermo Fisher, catalog number: J63122.AD)
PEG 4000 (Polysciences, catalog number: 16861-250)
Chlorimuron ethyl (Sulfonylurea) (Thermo Fisher, catalog number: J66605.03)
Hygromycin B (Thermo Fisher, catalog number: 10687010)
Penicillin/Streptomycin/Neomycin (PSN) antibiotic 100× solution (Thermo Fisher, catalog number: 15640055)
Q5 DNA polymerase (NEB, catalog number: M0491S)
PowerPol 2× PCR polymerase mix (Abclonal, catalog number: RK20718)
Solutions
20× (+) NO3 salts (see Recipes)
BDCM bottom media (see Recipes)
BDCM top media (see Recipes)
Liquid CM (see Recipes)
CM bottom media (see Recipes)
CM top media (see Recipes)
Trace elements (see Recipes)
Vitamin solution (see Recipes)
DNA extraction buffer (see Recipes)
70% ethanol (see Recipes)
Osmotic (OM) buffer (see Recipes)
ST buffer (see Recipes)
STC buffer (see Recipes)
PTC buffer (see Recipes)
1 M NaPO4 (pH 5.8) (see Recipes)
Recipes
20× (+) NO3 salts (1 L)
Reagent Quantity Sodium nitrate 120.0 g Potassium chloride 10.4 g Magnesium sulfate 10.4 g Potassium phosphate 30.4 g H2O Up to 1 L Autoclave at 121 °C for 25 min, cool to room temperature, then store at 4 °C.
BDCM bottom (1 L)
Reagent Quantity Yeast nitrogen base w/o amino acids 1.7 g Ammonium nitrate 2.0 g L-Asparagine 1.0 g Dextrose 10.0 g Sucrose 273.83 g H2O Up to 1 L Agar 1.5 g/100 mL Adjust pH to 6.0 with 1 M Na2HPO4 and autoclave at 121 °C for 25 min.
BDCM top (1 L)
Reagent Quantity Yeast nitrogen base w/o amino acids 1.7 g Ammonium nitrate 2.0 g L-Asparagine 1.0 g Dextrose 10.0 g H2O Up to 1 L Agar 1.0 g/100 mL Adjust pH to 6.0 with 1 M Na2HPO4 and autoclave at 121 °C for 25 min.
Liquid CM (1 L)
Reagent Quantity 20× (+) NO3 salts 50.0 mL Trace elements 1.0 mL Vitamin solution 1.0 mL Peptone 2.0 g Yeast extract 1.0 g Casamino acids 1.0 g Dextrose 10.0 g H2O Up to 1 L Adjust pH to 6.5 with 1 M NaOH and autoclave at 121 °C for 25 min.
CM bottom (1 L)
Reagent Quantity 20× (+) NO3 salts 50.0 mL Trace elements 1.0 mL Vitamin solution 1.0 mL Peptone 2.0 g Yeast extract 1.0 g Casamino acids 1.0 g Dextrose 10.0 g Sucrose 273.83 g H2O Up to 1 L Agar 1.5 g/100 mL Adjust pH to 6.5 with 1 M NaOH and autoclave at 121 °C for 25 min.
CM top (1 L)
Reagent Quantity 20× (+) NO3 salts 50.0 mL Trace elements 1.0 mL Vitamin solution 1.0 mL Peptone 2.0 g Yeast extract 1.0 g Casamino acids 1.0 g Dextrose 10.0 g H2O Up to 1 L Agar 1.0 g/100 mL Adjust pH to 6.5 with 1 M NaOH and autoclave at 121 °C for 25 min.
Trace elements (100 mL)
Reagent Quantity Sterile, autoclaved H2O 80 mL Zinc sulfate 2.2 g Boric acid 1.1 g Manganese chloride 0.5 g Iron sulfate 0.5 g Cobalt chloride hexahydrate 0.17 g Copper sulfate pentahydrate 0.16 g Sodium molybdate 0.15 g Na4EDTA 5.0 g Add all reagents in order. Using a hot plate, stir the solution until it comes to a boil. Cool to 60 °C and adjust the pH to 6.5 with 1 M KOH. Cool to room temperature. Adjust volume to 100 mL with ddH2O. Place in autoclaved bottles. Store at 4 °C. The solution should be a deep violet color before use.
Vitamin solution (100 mL)
Reagent Quantity Biotin 0.01 g Pyridoxin 0.01 g Thiamine 0.01 g Riboflavin 0.01 g PABA (p-aminobenzoic acid) 0.01 g Nicotinic acid 0.01 g Sterile, autoclaved H2O Up to 100 mL Place in autoclaved bottles. Store in the dark at 4 °C.
DNA extraction buffer (100 mL)
Reagent Quantity Potassium chloride 7.45 g 1 M Tris-HCl (pH 8.0) 10 mL 0.5 M EDTA (pH 8.0) 2 mL H2O Up to 100 mL Filter sterilize for long-term storage.
70% ethanol (1 L)
Reagent Final concentration Quantity Ethanol (absolute) 70% 700 mL ddH2O n/a 300 mL Total n/a 1000 mL Osmotic (OM) buffer (100 mL)
Reagent Quantity Magnesium sulfate heptahydrate 29.0 g 1 M NaPO4 (pH 5.8) 1 mL Lysing enzymes from Trichoderma 0.75 g H2O Up to 100 mL Adjust pH to 5.5 with 1 M Na2HPO4. Filter sterilize using a 0.45 μm syringe filter into 50 mL centrifuge tubes. Two 50 mL tubes should have 40 mL of OM buffer, while a third 50 mL tube should contain the remaining 20 mL. OM should be prepared the day before using and placed at 4 °C overnight.
ST buffer (500 mL)
Reagent Quantity Sorbitol 54.66 g 1 M Tris-HCl (pH 7.0) 50 mL H2O Up to 500 mL Autoclave for 25 min at 121 °C and store at 4 °C.
STC buffer (500 mL)
Reagent Quantity Sorbitol 109.32 g 1 M Tris-HCl (pH 7.5) 5.0 mL 1 M CaCl2 5.0 mL H2O Up to 500 mL Autoclave for 25 min at 121 °C and store at 4 °C.
PTC buffer (20 mL)
Reagent Quantity PEG 4000 8.0 g Sorbitol 3.64 g 1 M Tris-HCl (pH 7.5) 200 μL 1 M CaCl2 200 μL H2O Up to 20 mL Autoclave for 25 min at 121 °C and store at room temperature.
1 M NaPO4 (pH 5.8)
Reagent Amount 1 M NaH2PO4 92.1 mL H2O Up to 100 mL Adjust pH to 5.8 with 1 M Na2HPO4. Bring volume up to 100 mL and autoclave for 25 min at 121 °C.
Laboratory supplies
Pipettes 1,000 (Sartorius, catalog number: 14532053), 10 (Sartorius, catalog number: 11014473), and 200 μL (Sartorius, catalog number: 10195873)
Serological pipette (Scilogex SCI-Pet, Amazon)
25 mL serological pipette (Genesee Scientific, catalog number: 12-106)
Hemocytometer (Fisher Scientific, catalog number: 02-671-5)
Petri dishes (Fisher Scientific, catalog number: FB0875713)
Scalpel (Feather, catalog number: 02058370)
Miracloth (Calbiochem, catalog number: B36658)
Oakridge tube (Thermo Fisher, catalog number: 3119-0050)
50 mL conical tubes (Corning, catalog number: 430291)
500 mL flasks (Pyrex, catalog number: 4980)
Sterile paper towels (Scott multifold paper towels, Amazon)
Sterile funnel (Thermo Fisher, catalog number: 4252-0065PK)
100 mL bottles (Kimax, catalog number: 14397)
Dark trays (Mr. Pen plant trays, Amazon)
Aluminum foil (Reynolds Wrap, Amazon)
Incubator (Precision, catalog number: 31483)
1.5 mL microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-130)
0.45 μm syringe filters (Fisher Scientific, catalog number: 09-720-514)
Monarch PCR purification kit (NEB, catalog number: T1030S)
Equipment
Incubator (Percival, model: I36VL)
Centrifuge (Eppendorf, model: Centrifuge 5810 R with S-4-104 rotor)
Water bath (Thermo Fisher, model: 180 Water Bath Series)
Two-speed Waring laboratory blender (Fisher Scientific, catalog number: 7010S)
Eberbach container for Waring blenders (Fisher Scientific, catalog number: 14-509-25)
Incu-shaker (Benchmark, model: 10LR)
Thermal cycler (Bio-Rad, model: C1000 Touch)
Vacufuge plus (Eppendorf, model: 022820109)
Centrifuge for Eppendorf tubes (Eppendorf, model: Centrifuge 5418)
Homogenizer (Amazon, SP Bel-Art ProCulture F65100-0000)
Hot plate and stirrer (Thermo Fisher, catalog number: SP88854100)
Procedure
Primer design and PCR reactions
Generate upstream and downstream regions outside of gene of interest’s open reading frame and selectable marker pieces.
Design primers that amplify 1 kb upstream of the beginning of your gene’s open reading frame (L1, L2; Figure 1).
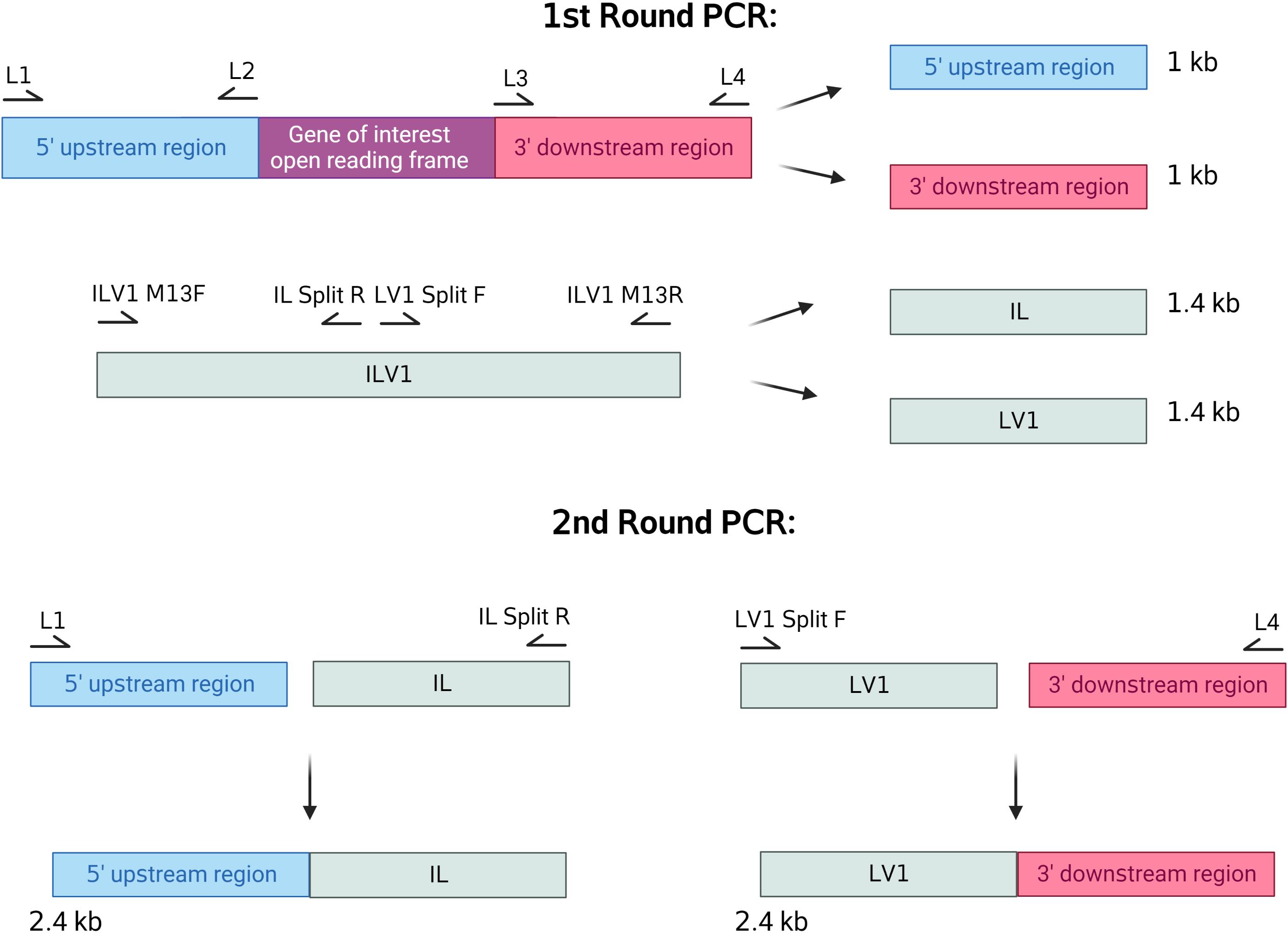
Figure 1. Split marker primer designDesign primers that amplify 1 kb downstream following the end of your gene’s open reading frame (L3, L4).
Refer to Table 1 to see the primers to amplify the two pieces of the ILV1 gene that confers resistance to sulfonylurea (see Note 1).
Table 1. List of oligonucleotides used in the ILV1 transformation (see Note 2)
ID 5′----3′ sequences ILV1 gene ILV1 M13F CGCCAGGGTTTTCCCAGTCACGAC GTCGACGTGCCAACGCCACAG IL Split R AAGCATGTGCAGTGCCTTC LV1-M13R AGCGGATAACAATTTCACACAGGA TAGGTCGACGTGAGAGCATGC LV1Split F GAGGCCGACGTCATAGGCATC M13 sequences for the selectable marker M13F CGCCAGGGGTTTTCCCAGTCACGAC M13R AGCGGATAACAATTTCACACAGGA M13 for the gene of interest M13F TCCTGTGTGAAATTGTTATCCGCT M13R GTCGTGACTGGGAAAACCCTGGCG
Amplify flanks using PCR.
Amplify the upstream and downstream region of your gene of interest using M. oryzae genomic DNA as the template.
Amplify the two pieces of your selectable marker using an ILV1 containing plasmid as the template.
Purify the amplified fragments using a PCR purification kit.
Fuse the upstream region of your gene of interest with the 5′ end of the selectable marker together using the 5′ products generated from the first round of PCR. Final DNA concentration should be approximately 5 μg (see Note 3).
Fuse the downstream region of your gene of interest with the 3′ end of the selectable marker together using the 3′ products generated from the first round of PCR. Final DNA concentration should be approximately 5 μg.
Purify the resulting fused fragments using a PCR purification kit.
Combine the fused fragment containing the upstream region of the gene and the 5′ end of the selectable marker along with the downstream region of the gene and 3′ end of the selectable marker in one tube.
Dry each tube until only ~10 μL of DNA are present using a vacuum concentrator.
Add 50 μL of STC buffer to each tube (see Note 4).
Protoplast generation
Grow M. oryzae mycelia on liquid complete medium (CM) (Figure 2).

Figure 2. Materials needed for generation of liquid CM mycelial culturePlate fungal strain on three CM plates each containing three colonies. M. oryzae was grown at 24 °C with a 12 h photoperiod using a I36VL Percival incubator with a IncuWhite Linear LED lighting system, with an intensity of 120 μmol/m2/s (Figure 3).
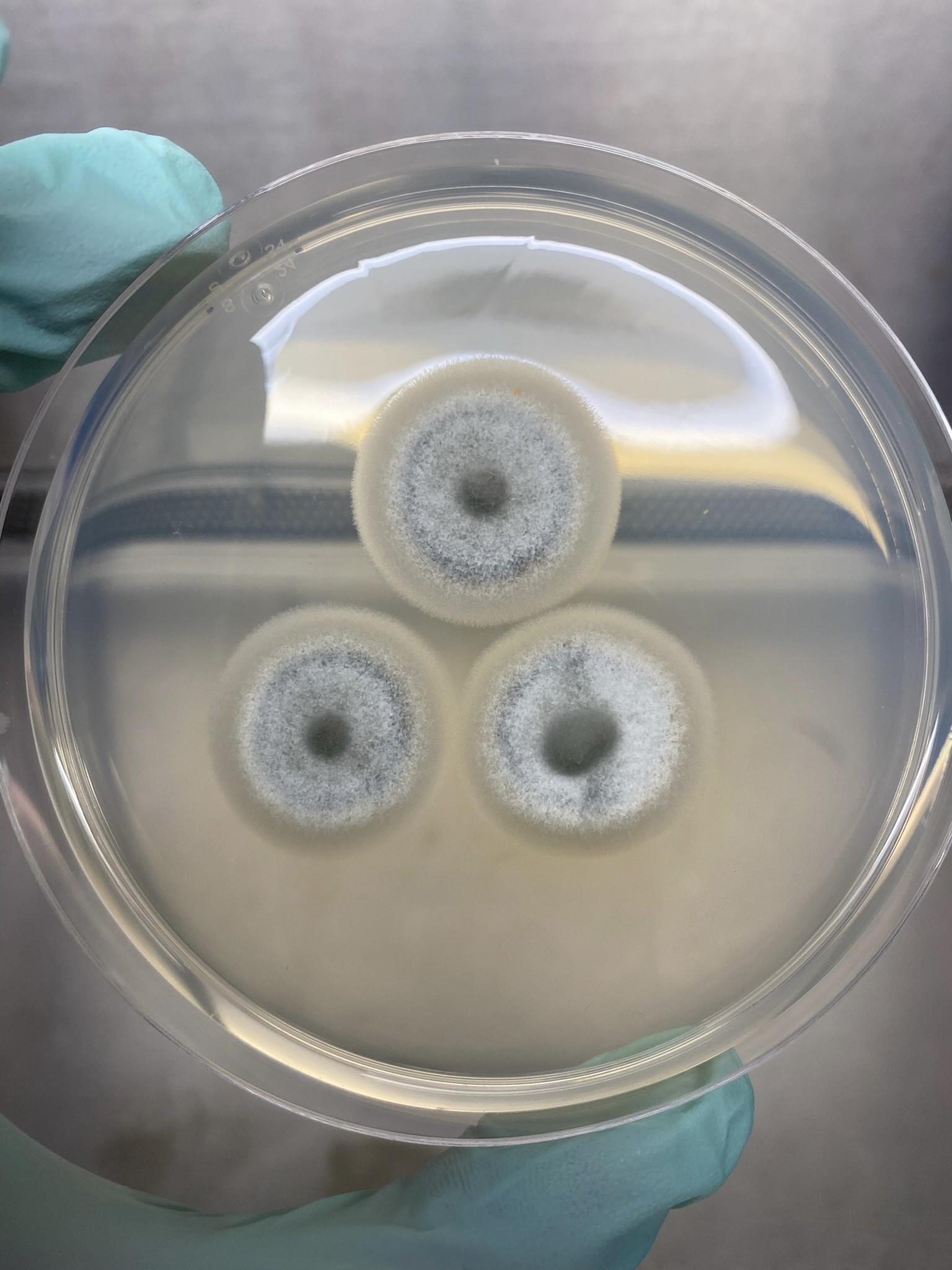
Figure 3. CM plate containing three 5-day-old coloniesAfter five days, cut the growing circumference of mycelia from each colony using a sterile scalpel. (Video 1).
 Video 1. Cutting growing circumference of mycelia. Using a blade, the growing edge of mycelia is cut from CM plates and placed in liquid CM to blend.
Video 1. Cutting growing circumference of mycelia. Using a blade, the growing edge of mycelia is cut from CM plates and placed in liquid CM to blend.Combine the cut edges of mycelium in 250 mL of liquid CM in a sterile blender. Blend on high for 10 s, then on low for 10 s.
Transfer blended mycelium into a 500 mL flask and add 250 μL of antibiotic solution to the culture. The PSN antibiotic solution from Thermo Fisher is used at a 1× concentration.
Incubate culture at 24 °C with a 12 h photoperiod for approximately 48 h with shaking at 150 rpm (see Note 5).
After 24 h, add an additional 100 mL of liquid CM to the culture along with 100 μL of antibiotic solution and leave in shaking incubator at 200 rpm for the remainder of the 48 h.
Lyse mycelia to generate protoplast.
Filter mycelia from liquid culture using a sterile funnel and sterile Miracloth (Figures 4 and 5).

Figure 4. Materials needed to filter fungal culture
Figure 5. Filtered myceliaRinse filtered mycelia with sterile distilled water.
Using sterile paper towels, dry the filtered mycelia by squeezing the mycelia between paper towels to remove as much moisture as possible.
Divide dry mycelia into two 50 mL falcon tubes containing 40 mL of OM buffer.
Incubate tubes at 29 °C with shaking at 75 rpm for 3 h in a shaker incubator (see Note 6).
Harvest protoplast; do all of the following on ice.
Divide each falcon tube among two sterile clear polycarbonate Oakridge tubes.
Slowly add cold ST buffer drop by drop until each Oakridge tube is filled to the neck (see Note 7) (Video 2).
Video 2. Slow addition of ST buffer to Oakridge tube. Following mycelia lysis, the OM buffer and mycelia mixture is divided between two Oakridge tubes. Using a serological pipette, cold ST buffer is added slowly until the tube is filled to the neck.Centrifuge tubes at 3,200× g for 25 min at 4 °C using a swinging bucket centrifuge with brakes disabled to ensure proper generation of protoplast layer.
Using a 1 mL pipette, slowly draw up protoplasts from each tube into new Oakridge tubes. Protoplasts from two Oakridge tubes can be combined into one new Oakridge tube (see Note 8) (Figure 6).

Figure 6. Protoplast appearance in Oakridge tube after centrifugingAdd STC buffer to the neck of each new Oakridge tube containing protoplasts.
Centrifuge tubes at 1,900× g for 10 min at 4 °C, with breaks half engaged.
A protoplast pellet should now be seen.
Washing protoplast.
Decant all STC buffer from each Oakridge tube.
Gently resuspend pellet with 5 mL of cold STC buffer.
Two resuspended pellets can be combined into one Oakridge tube.
Add STC buffer to the neck of each Oakridge tube.
Centrifuge tubes at 1,900× g for 10 min at 4 °C, with breaks half engaged.
Repeat steps B4a–B4e 2 additional times for three total protoplast washes.
Protoplast transformation.
Resuspend final protoplast pellet in 1 mL of STC buffer.
Using a hemocytometer, calculate protoplast concentration. A proper yield should be 2 × 108–5 × 108 protoplasts. Concentrate or dilute protoplasts with STC accordingly (Figure 7).

Figure 7. Protoplast after harvest. Visualization is at 40× magnification.For each transformation, in a 1.5 mL tube, add 100 μL of protoplast to DNA (5 μg of each flank) that has been combined and resuspended in 50 μL of STC (see Note 9).
Incubate tubes at room temperature for 20 min (see Note 10).
*Please note that this is a time critical step. Avoid leaving the protoplasts in PTC for longer than the allotted time. Add 500 μL of PTC buffer to each tube. After adding PTC to the final tube, begin a 15 min timer (see Note 11).
Repeat the addition of 500 μL of PTC buffer to each tube until all tubes have a total of 1 mL of PTC.
Invert the tubes periodically during the 15 min incubation period.
Divide and label Petri plates. Four plates should be used for each strain (Figure 8).

Figure 8. Four Petri plates set aside per strain
Protoplast plating.
Add protoplasts to 100 mL of bottom media. *Please ensure that the bottom media is at approximately 45 °C as hotter temperatures will destroy the protoplast. Gently swirl to mix (see Notes 12 and 13).
Pour and divide media between four Petri dishes (see Note 14) (Video 3).
Video 3. Division of media. Following PTC incubation, the protoplasts are added to barely warm bottom media, swirled, and plated between four Petri dishes.Once solidified, transfer plates to clean, dark trays and cover with aluminum foil.
Incubate plates at 24 °C for 16 h.
The next day, add top media with corresponding antibiotics to transformation plates (see Note 15).
Place plates back in dark trays and incubate for at least five days until colonies appear (Figure 9).
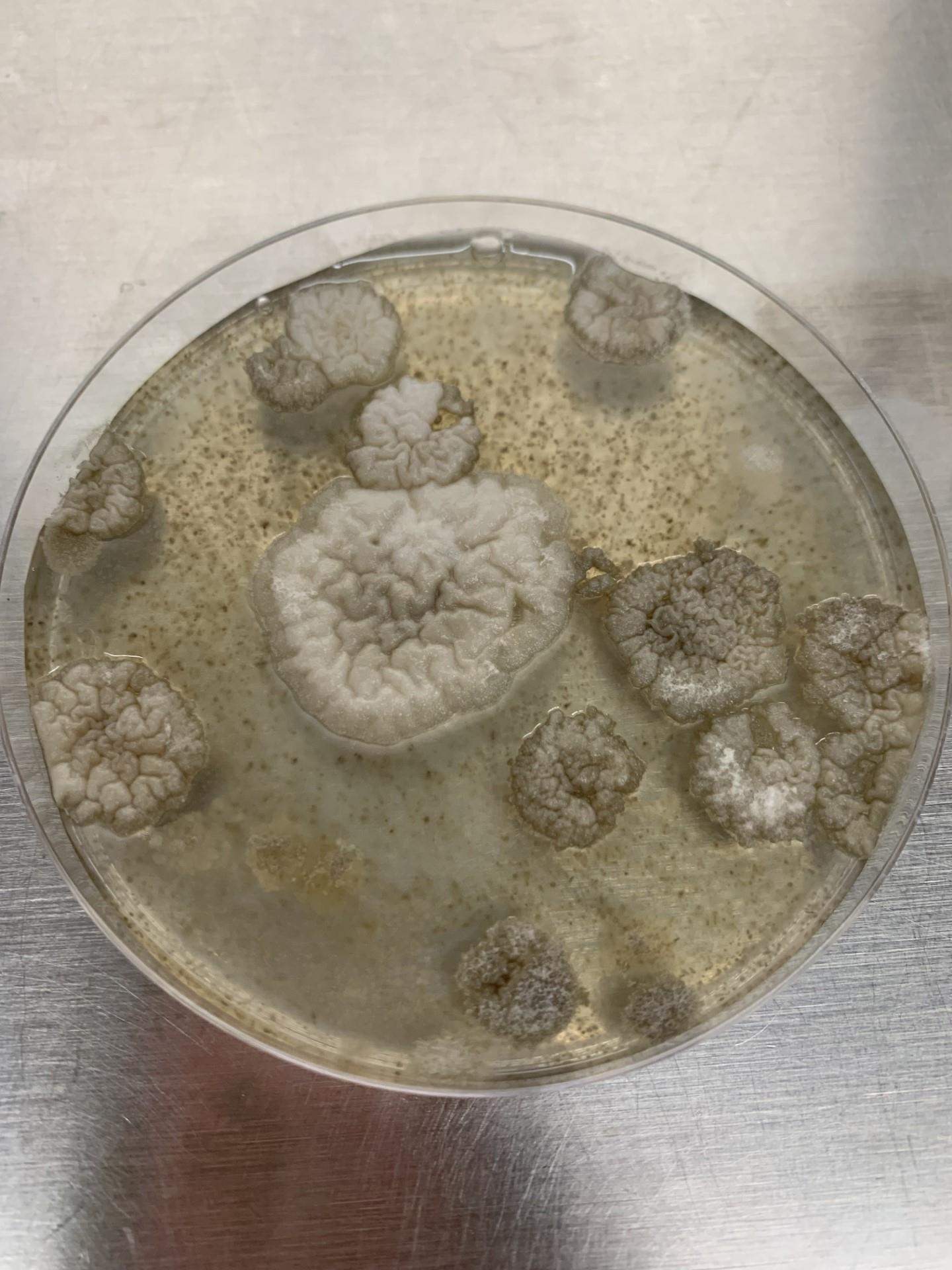
Figure 9. Colonies after fungal transformation
Screening fungal colonies
DNA extraction.
When colonies appear on the top layer of transformation plates, re-plate colonies in a grid on new media with corresponding antibiotics.
After 3–6 days, transfer a small amount of mycelia using a sterile toothpick or pipette tip from each colony into a sterile 1.7 mL tube and label each colony accordingly.
To each tube, add 500 μL of DNA extraction buffer.
Using an electronic homogenizer, pulverize mycelia for ~10 s (see Note 16).
Spin tubes at 2,100× g for 10 min at room temperature.
Following centrifugation, transfer the supernatant to a new tube containing 300 μL of 100% isopropanol. Mix tube (see Note 17).
Centrifuge tubes at 12,000× g for 10 min at room temperature.
Discard as much supernatant as possible.
Wash tube with 300 μL of 70% ethanol.
Centrifuge tubes at 12,000× g for 10 min at room temperature.
Discard as much supernatant as possible.
Air dry tubes by placing inverted tubes on a tissue for ~15 min.
After all ethanol has evaporated, add 30 μL of sterile water and 1 μL of RNase to each tube.
Incubate tube for 20 min at room temperature.
Incubate tube at 55 °C for 15 min to inactivate RNase.
Following incubation, add 150 μL of sterile water to each tube. Mix.
Use 1 μL of DNA extraction for screening.
PCR verification.
Using primers that fall outside the L1 and L4 region, run a PCR reaction for each of the potential mutants using the DNA extraction as a template. Use the wild-type Guy11 as a negative control (see Notes 18 and 19).
Run PCR amplification on a gel.
Verify band size for confirmation of positive transformation (see Note 20) (Figure 10).
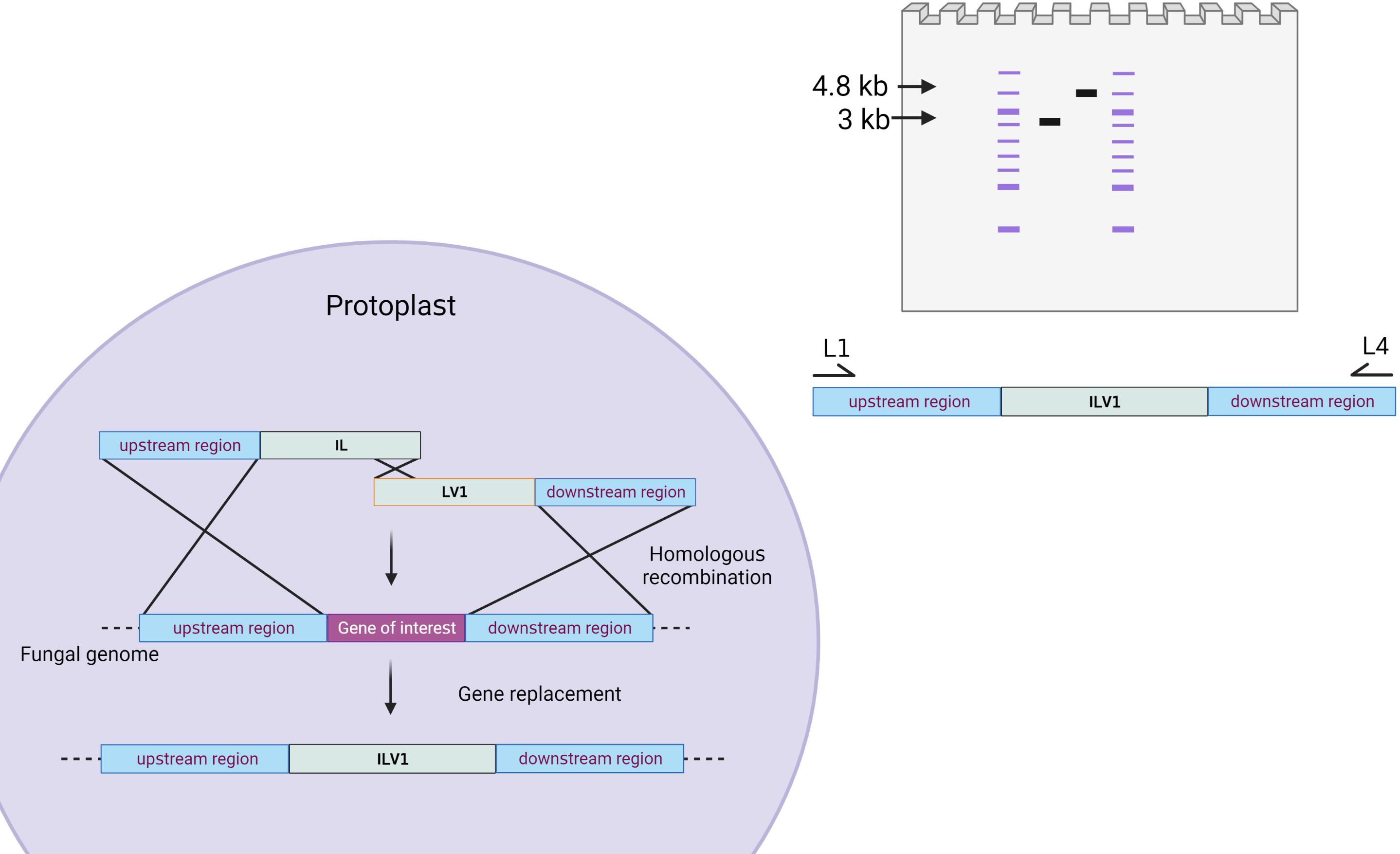
Figure 10. Schematic of DNA gel depicting arbitrary amplifications following transformation. This arbitrary gene of interest is proposed to be 1 kb (although sizes will vary based on the gene of interest selected). A wild-type band would include the 1 kb upstream region, the 1 kb gene, and the 1 kb downstream region, yielding a size of 3 kb. A positive mutant would carry a higher band size at 4.8 kb, including the 1 kb upstream region, the 2.8 kb ILV1 gene, and the 1 kb downstream region. Please note that these sizes will vary based on the size difference between the gene of interest and the selectable marker.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Fernandez et al. (2021). Role of two metacaspases in development and pathogenicity of the rice blast fungus Magnaporthe oryzae. mBio (Figure S1, panel c). Mutant strains for M. oryzae metacaspases were generated using this protocol.
General notes and troubleshooting
General notes
This protocol details the use of the ILV1 gene that confers resistance to sulfonylurea (gene size: 2.8 kb). However, other selectable markers can be used, such as hygromycin. For these PCR reactions, it is recommended to use a high-fidelity DNA polymerase, such as Q5 and Phusion.
Please note that certain primers require the addition of M13 primer sequences. The M13 sequences promote the fusion of fragments generated during the first round of PCR.
Typically, a PCR reaction volume of 100–200 μL is required to generate 5 μg of PCR product.
For transforming plasmids, 2–5 μg of plasmid should be linearized with a restriction enzyme before fungal transformation, PCR purified, dried to ~10 μL, and resuspended in 50 μL of STC buffer.
Please note that a healthy growth following this 48-h incubation period will produce a light brown media color. An overgrown culture will produce dark brown/black media.
During this incubation period, place bottles containing bottom medium in 55 °C water bath. Also, ensure centrifuge is at 4 °C.
Ensure that the addition of ST buffer to the Oakridge tubes is done drop by drop to ensure proper protoplast harvest.
The protoplasts are located at the hazy interface towards the middle of the tube. Ensure that when pipetting up protoplasts, the mycelium on the bottom is undisturbed.
A control should be included that has only 50 μL of STC buffer with no DNA and combined with 100 μL of protoplasts to check for protoplast integrity.
At this point, begin cooling the bottom medium bottles from 55 °C. By the end of the PTC buffer incubation, the media should be cool enough to touch with a gloved hand and should be around 45 °C.
Begin by pipetting the PTC buffer around the side of the tube until all PTC has been added. Slowly pipette the PTC buffer and protoplast mixture up twice. After mixing, gently invert the tubes twice.
Ensure that the media corresponds to the specific selection marker used. CM media is used for hygromycin selection; BDCM media is used for sulfonylurea selection; BASTA media is used for BASTA selection. Hygromycin is used as a 50 mg/mL stock concentration to a final concentration of 100 μg/μL. Sulfonylurea is used as a 100 mg/mL stock concentration to a final concentration of 50 μg/μL. BASTA is used as a 50 mg/mL stock concentration to a final concentration of 100 μg/μL.
Bottom media should be aliquoted in 100 mL volumes per transformation before autoclaving.
Ensure that each plate is 1/3 filled with media. Adding too much media to each plate will prevent properly adding top media the next day.
Bottom media should be used to plate protoplasts initially. Make sure that the top media is used for selection the following day.
The ground mycelia should not be left in the extraction buffer for a prolonged period of time. Additionally, if extracting more than one sample, the homogenizing pestle can be disinfected between samples by dipping in sterile water, followed by 100% ethanol.
Following this step, the samples can be left at -20 °C overnight and the DNA extraction can continue the next day.
Confirmation primers that fall outside the L1 and L4 region will ensure that the gene replacement has an endogenous incorporation of the selectable marker. Additionally, confirmation primers can be paired with internal split marker primers to yield smaller PCR fragments that can also be validated based on fragment size.
For this PCR screen, DNA polymerases such as DreamTaq from Thermo Fisher or PowerPol from ABclonal may be used for a more inexpensive reaction.
Amplicons of potential positive transformants can be confirmed using a sequencing service. Additionally, positive transformants can be further validated using Southern blot analysis.
Acknowledgments
This work was supported by the Early Career Grant and Undergraduate Research Internship program from UF/IFAS research. Graphical overview and Figures 1 and 10 were created with BioRender.com.
Competing interests
There are no conflicts of interest or competing interests.
References
- Asibi, A. E., Chai, Q. and Coulter, J. A. (2019). Rice blast: A disease with implications for global food security. Agronomy 9(8): 451.
- Fernandez, J., Lopez, V., Kinch, L., Pfeifer, M. A., Gray, H., Garcia, N., Grishin, N. V., Khang, C. H. and Orth, K. (2021). Role of two metacaspases in development and pathogenicity of the fungus Magnaporthe oryzae. mBio 12(1): e03471-20.
- Fernandez, J., Marroquin-Guzman, M., Nandakumar, R., Shijo, S., Cornwell, K. M., Li, G. and Wilson, R. A. (2014a). Plant defence suppression is mediated by a fungal sirtuin during rice infection by Magnaporthe oryzae. Mol. Microbiol. 94(1): 70–88.
- Fernandez, J., Marroquin-Guzman, M. and Wilson, R. A. (2014b). Evidence for a transketolase-mediated metabolic checkpoint governing biotrophic growth in rice cells by the blast fungus Magnaporthe oryzae. PLoS Pathog. 10(9): e1004354.
- Fernandez, J. and Orth, K. (2018). Rise of a cereal killer: The biology of Magnaporthe oryzae biotrophic growth. Trends Microbiol. 26(7): 582–597.
- Foster, A. J., Martin-Urdiroz, M., Yan, X., Wright, H. S., Soanes, D. M. and Talbot, N. J. (2018). CRISPR-Cas9 ribonucleoprotein-mediated co-editing and counterselection in the rice blast fungus. Sci. Rep. 8(1): e1038/s41598-018-32702-w.
- Sweigard, J. A., Carroll, A. M., Kang, S., Farrall, L., Chumley, F. G. and Valent, B. (1995). Identification, cloning, and characterization of PWL2, a gene for host species specificity in the rice blast fungus. Plant Cell 7(8): 1221–1233.
Article Information
Publication history
Published: Sep 5, 2023
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Garcia, N., Farmer, A., Baptiste, R. and Fernandez, J. (2023). Gene Replacement by a Selectable Marker in the Filamentous Fungus Magnaporthe oryzae. Bio-protocol 13(17): e4809. DOI: 10.21769/BioProtoc.4809.
Category
Microbiology > Microbe-host interactions > Fungus
Molecular Biology > DNA > Transformation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.
![]() Tips for asking effective questions
Tips for asking effective questions
+ Description
Write a detailed description. Include all information that will help others answer your question including experimental processes, conditions, and relevant images.